ACS PRF | ACS
All e-Annual Reports

42428-G3
Electrochemical Studies of the Multicopper Oxidase CueO from E.Coli
In this project we have examined the bacterial multicopper oxidase CueO from E. coli using the electrochemical technique of protein film voltammetry (PFV). CueO is a multicopper oxidase (MCO) that couples catalytic reduction of dioxygen by four electrons (forming water), to the oxidation of either aromatic molecules, or metal ions. We have investigated CueO in the following three aims:
1. Assess the reduction potentials of CueO copper ions,
2. Measure CueO electron transfer kinetics, and
3. Determine redox-based properties of CueO catalysis.
In the first year of this project, Aims 1 and 2 were primarily addressed through direct PFV experiements. In the second year of this project, Aim 3 has been the primary focus.
Aim 3. Determine redox-based properties of CueO catalysis
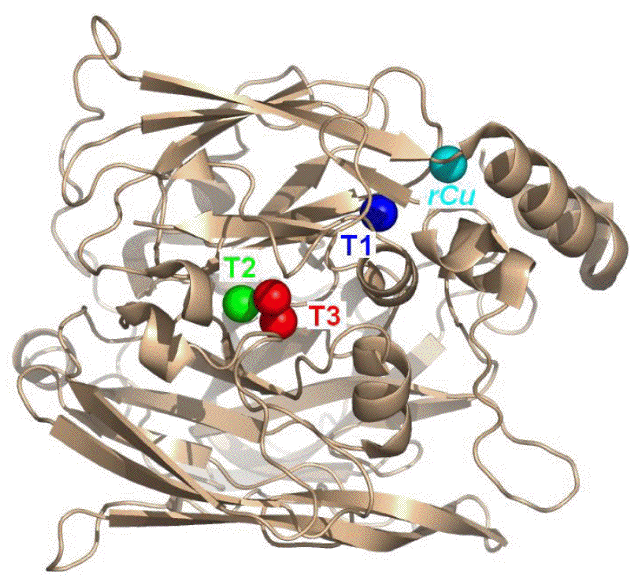
Figure 1. Crystal structure of CueO, showing the positions of the T1, T2 and T3 copper centers.
Previously, we had found that CueO could be "wired" to either graphite- or gold-based electrodes, to effect the catalytic reduction of oxygen to water at relatively high potentials. The achieve this, 4 electrons must be taken up at the CueO active site comprised of the Type 2 and Type 3 copper ions (T2 and T3, in Figure 1), where electron transfer occurs via the Type 1 center (blue, in Fig 1). The T1 center therefore acts as an essential electron transfer component, much like a relay, for specific flow of electrons coupled to catalysis. In this Aim, we have most recently examined the role of the T1 center to modulate or to govern the electrocatalytic properties of adsorbed CueO.
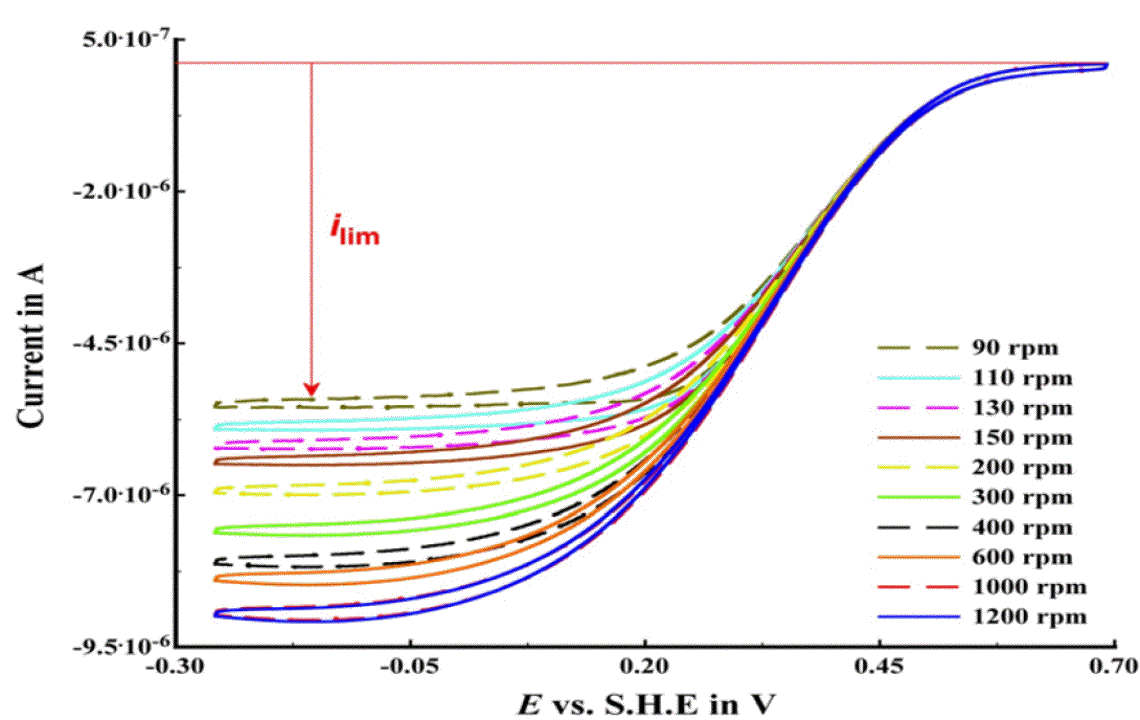
Figure 2. Catalytic PFV (red) for CueO immobilized on p-(MBT)-modified gold.
To determine that CueO at the electrode behaves like native enzyme, we have conducted PFV in the presence of the substrate, dioxygen. Fig 2 shows that CueO, at a rotating disc electrode. Here, rapid reduction of dioxygen occurs as the potential is scanned from positive to negative values, indicating that substrate turnover is fast on the time scale of the electrochemical experiment. Importantly, we have shown that we can defeat the limitation of substrate diffusion to electrode surface. The limiting current observed grows, as the rotation rate of the electrode is increased from 90 rpm to 1000 rpm. At any value greater than 1000 rpm, the observed sigmoidal waveshape is identical, indicating that the system is under "catalytic control", i.e., that the enzyme is determining the magnitude of the limiting current, and not the diffusion of substrate molecules. This result has not been achieved previously in the literature.
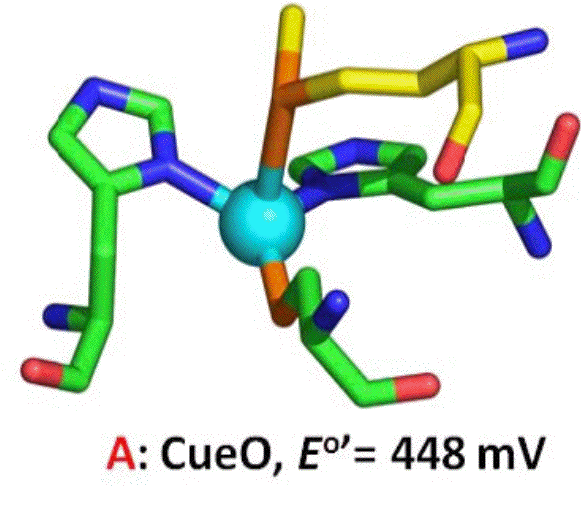
Figure 3. The T1 copper center of CueO.
The shape of the resulting voltammograms are highly symmetric in the forward and reverse directions, indicating that the system is engaging in fast electron transfer that enables catalysis to occur with no substrate depletion. However, the potential region covered by the sigmoidal wave is substantial, over 400 mV. We hypothesize that the broad shape of the data shown in Figure 2 indicates that the Type 1 center (Fig 3) serves as a gateway for subsequent, rate-determining electron transfers into the active site of CueO. Using direct spectrophotometric titration of T1, we have found the midpoint potential to be 448 mV, placing the resting state T1 potential at the onset of the catalytic wave. Thus, at lower values of potential, the T1 center is still being reduced, and handing-off (relaying) electrons to the active site. To support this model, we have designed, produced and characterized mutants of the Type 1 center to show that as the T1 potential changes, so will the on-set of electrocatalysis.
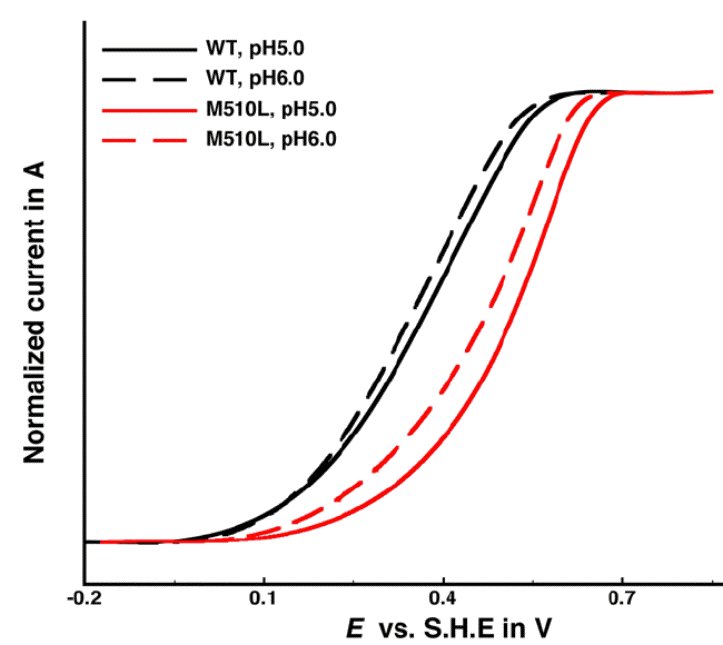
Figure 4. Catalytic voltammetry of M510L CueO, compared to wild-type. Ca Ca
We have re-cloned CueO and used this new clone to prepare site-directed mutants of the T1 center that should posses a higher reduction potential. By removing the Met510 ligand (Fig 3), we hoped to stabilize the CuI oxidation state, and increase the potential. Thus, the M510L mutant was constructed, expressed and produced in ample quantities. Spectrophotometric titration of the T1 center showed that the new T1 midpoint potential was indeed higher by ~130 mV higher than the wild-type protein (574 mV). Catalytic voltammetry was again used (Fig 4), revealing that in support of our model, the same broad waves are generated, but with an onset potential >150mV greater than wild-type.
Thus, our model of T1 acting as an electrochemical relay has been supported, and we believe that in future work, we can tune the CueO T1 potential to even greater values. This will enable CueO to be a high-potential dioxygen-reducing cathode as a component in a biological fuel cell.