
ACS PRF | ACS
All e-Annual Reports

45485-AC5
Multi-Scale Simulation Studies of Anhydrous Proton-Conducting Materials for Use in High-Temperature Fuel Cell Applications
Project 1: Connecting microscopic proton transfer mechanisms to proton transport kinetics in hydrogen-bonded networks
Proton transfer (PT), including proton holes, and the migration of positive and negative topological charge defects in hydrogen--bonded (HB) networks, is central to a myriad of processes in chemistry, physics, materials science, and biology. Although simulations 1,2,3 have elucidated many details of such structural diffusion mechanisms, a theory connecting a particular defect's solvation structure and microscopic PT mechanism to macroscopic charge transport kinetics is lacking. The PI introduced a general statistical-mechanical framework that achieves the aforementioned goal within a chemical master equation (CME) approach4. The theory employs the concept of population correlation functions5 and associated rate equations6 as commonly applied to HB kinetics, but extends these essential ideas in key ways.
Suppose the HB donors/acceptors (A) are identical, i.e. A—H……..A. Addition or removal of a proton creates a cationic or anionic defect site A*, e.g. H3O+ or OH- Define two charge defect population functions: h(t)=1 if a tagged A atom is A* at time t (h(t)=0 otherwise), whereas H(t)=1 if A* retains its identity continuously up to time t. These functions describe topological defect patterns as opposed to their counterparts for HB kinetics in pure liquids5,6. Now introduce an intermittent PT correlation function CiAA (t)= <h(0)h(t)>/<h> that yields the probability of finding the same A* at times t=0 and t, irrespective of an identity change in the interim and a continuous PT correlation function CcAA (t)=<h(0)H(t)> /<h> that does not allow for identity change of A* in [0,t].
Suppose there are p solvation patterns, each with a coordination number n1,…,np. Then, CcAA (t)=<h(0)H(t)> /<h> is particularized by introducing CcAA-ni(t) describing the probability of finding the same A* continuously up to time t given that its coordination number is ni at time t=0. Lifetimes are obtained from
![]()
which is the average time needed for PT from A* to a first solvation shell member and 1/τexch is the average PT rate, with an analogous definition of τexch,ni. `Proton rattling' events, in which the defect returns to its original A* site after two successive PTs, can be either included or excluded. Including allows comparison to femtosecond experiments7. Excluding them provides a clearer picture of the transport process.
The decay of CiAA (t)depends not only on 1/τexch but also on the reverse process. In order to disentangle the coupled forward/backward kinetics6, a nearest--neighbor PT correlation function CnnAA (t) is introduced, which measures the probability of finding an A atom in the hydration shell of A* at time t given that it was the A* site at t=0; an analogous definition holds for CnnAA-ni (t). Including the reverse reaction leads to the rate equation for PT
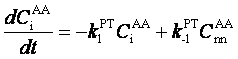
with k1PT and k-1PT being the forward and backward PT rate constants, respectively. This defines a general formalism, independent of the particular HB network that allows the kinetics of PT and structural diffusion to be investigated theoretically in a quantitative manner. Akin to other CME approaches9, the formalism permits one to connect long--time processes to specific microscopic motions. The theory was applied to the well-understood problem of hydronium diffusion in water, yielding time scales in agreement with those of ultrafast spectroscopy studies7 and was subsequently used to decide among three proposed mechanisms of hydroxide diffusion in water8.
Project 2: Development of force field parameters for imidazole attached to a polymeric spacer
We have now developed a set of force field parameters for the attachment of imidazole to a polyethyleneoxide spacer, as discussed in the original proposal. Starting parameters were developed from those of Krishnan and Balasubramanian10 for polyethyleneoxide and CHARMM22 for imidazole11. The figure and tables below show the atom names and the force field parameters, including charges and intramolecular potential parameters. All calculations were performed using Gaussian0312 at the Hartree-Fock level of theory with a 6-31G* basis set.
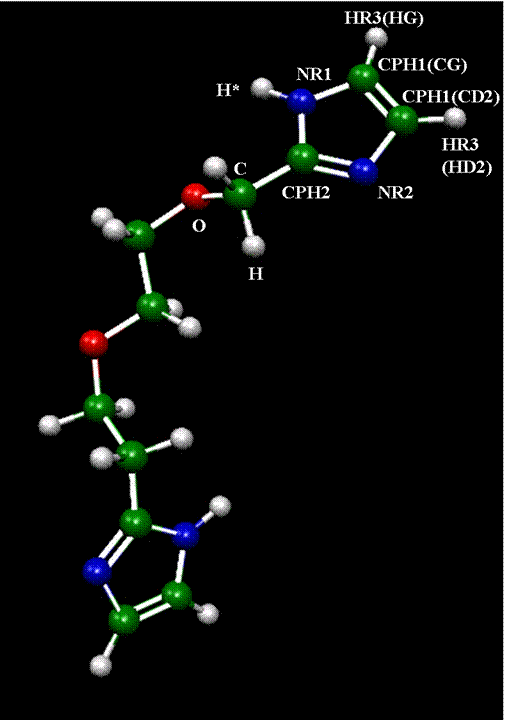

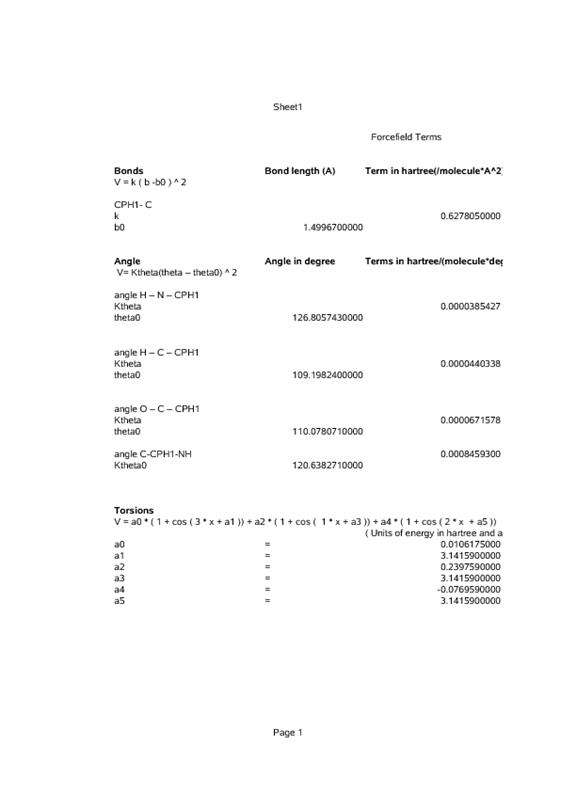
References:
- D. Marx, ChemPhysChem 7, 1848 (2006).
- G. A. Voth, Acc. Chem. Res. 39, 143 (2006).
- B. Roux, Acc. Chem. Res. 35, 366 (2002).
- A. Chandra, M. E. Tuckerman and D. Marx, Phys. Rev. Lett. (in press).
- F. H. Stillinger, Adv. Chem. Phys. 31, 1 (1975).
- A. Luzar and D.
- S. Woutersen and H. J. Bakker, Phys. Rev. Lett. 96, 138305 (2006).
- M. E. Tuckerman, A. Chandra and D. Marx, Acc. Chem. Res. 39, 151 (2006).
- J. D. Chodera, et al. J. Chem. Phys. 126, 155101 (2007).
- M. Krishnan and S. Balasubramanian. Chem. Phys. Lett. 385, 351 (2004).
- A. MacKerell, Jr., et al. J. Phys. Chem. B 102, 3586 (1998).
- M. J. Frisch, et al. Gaussian 03, revision C.02; Gaussian, Inc.: