
ACS PRF | ACS
All e-Annual Reports

43277-B5
Better Understanding the Behavior of Hexane Deposited onto Graphite Using Improved Computer Simulations
I. General Comments
We have been able to meet the proposed objectives regarding hexane (C6H14, or C6) on graphite (C6/gr). The PI appreciates that next summer is the last active summer for the grant and so he has applied for a sabbatical in 2008. It would give 8.5 continuous months for research and dissemination. At a teaching University such an opportunity would prove invaluable. We have presented results related to the above mentioned grant at various conferences 7 times, with student co – authors. Manuscripts are under review and in preparation.
II. Hexane on Graphite
As originally proposed, we have completed an extensive series of explicit – hydrogen simulations of hexane on graphite. The simulations address the system’s behavior at monolayer coverage as well as the submonolayer. We currently have one manuscript under review in Langmuir but it is comprehensive.
Specifically, we look at coverages in the range 0.5 ≤ ρ ≤ 1 monolayers. The dynamics seen in the fully-atomistic model agree well with experiment. Energetics and structural parameters are more reasonable and, collectively, the results from the simulations in this work demonstrate that the explicit-hydrogen model of hexane is substantially more realistic than the UA approximation.
At various densities the system has differing topologies, as shown in figure 1.
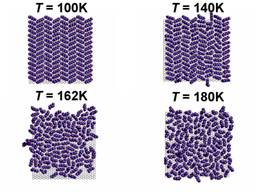

Figure 1. Snapshots of various phases of C6/gr ranging from the solid herringbone to the isotropic liquid (left panel). Snapshots of typical submonolayer topolgies (right panel).
III. Tetracosane on Graphite
I am very grateful to have received an SRF to bring a new collaborator from
After carefully varying NAMD simulation parameters we found that the electrostatic scaling parameter (freely adjustable) has a profound influence on molecular flexibility for longer molecules and hence internal dynamics. As the scaling factor is decreased, the molecules become stiffer and hence the transition temperature increases (figure 3). This is significant because we have determined an appropriate scaling factor which applies to all physisorbed alkane calculations and have done long (5 ns) simulations.
Manuscripts are in preparation regarding the scaling factor study as well as another regarding the 5 ns monolayer runs. We are going to simulate C24 bilayer, trilayer and submonolayer systems as well, with at least three more manuscripts envisioned. Adjusting the electrostatic scaling factor for C6 / gr made only about a 5K difference in melting temperature, due mainly to the inflexibility of C6.
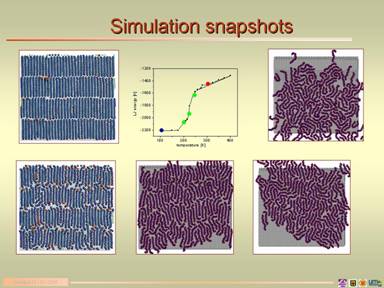
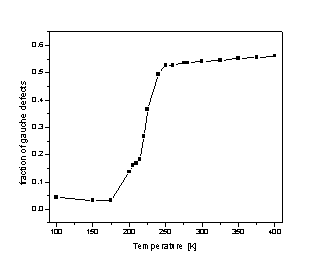
Figure 2. Plot of gauche (torsion) defects vs. temperature with full electrostatics (right panel). Snapshots of the C24/gr system in the solid phase through melting into the liquid (left panel). In the first two plots, a red color indicates increased elevation from the graphite surface. The graph in the middle is of interaction energy vs. temperature and shows the solid (blue), liquid (red) and melting transition region (green). The internal energy plot agrees with the gauche defect curve (right).
Figure 3. Snapshots of final configurations of C24/gr with various scaling factor values (SF). The profound influence of the scaling factor can be seen. A red color indicates increased elevation from the graphite surface. IV. Fullerenes on Graphite / Percolation We still intend to study percolation in surface systems, and are now in an excellent position to continue. While examining the electrostatic scaling issue for C24/gr, we became interested in C60 fullerenes on graphite. A collaborator (Renee Diehl – Figure 4. Final configuration snapshot of large scale percolating C60/gr (left) and calculated diffraction pattern (right).
