ACS PRF | ACS
All e-Annual Reports

44063-G3
Bridging the Gap Between Early and Late-Metal Catalysis: d2 Square-Planar Complexes That Mimic Their d8 Counterparts
Early (groups III–VI) and late (groups VII–X) transition metal complexes have distinct structural and reactivity properties. Early metals favor high oxidation states, high coordination numbers, are typically electrophilic and are intolerant of many functional groups. The opposite is true for late metal complexes, in which low coordinate and low oxidation states prevail and a wide range of functional groups are tolerated. Since initial investigations by Shaw, pincer-ligated metal complexes have rapidly emerged, with great success, as catalysts for a wide range of important reactions. Their broad utility is derived from an inherent modular architecture, thereby enabling precise control over both the steric and electronic coordination environment of metals. In addition, pincer ligands are covalently bound to metals via at least one metal-carbon σ–bond, which prevents ligand dissociation and renders many complexes thermally robust, often leading to high turnover numbers. Chemists quickly recognized the inherent stability and potential of the pincer architecture and have since systematically modified the pendant arms and backbone of the classic pincer skeleton seemingly at will. After 15 years of modification however, only two properties remain constant: (1) pincer ligands are monoanionic with few exceptions, and (2) they are used almost exclusively with late transition metals. The soft–hard–soft (1) donor atoms of classic pincer ligands are well suited for the softer late metals. To accommodate harder early transition metals we designed two hard–hard–hard (2, 3) pincer motifs (Figure 1).
Figure. 1. Classic monoanionic 1 vs. New trianionic 2 and 3 pincer ligands: SHS vs. HHH
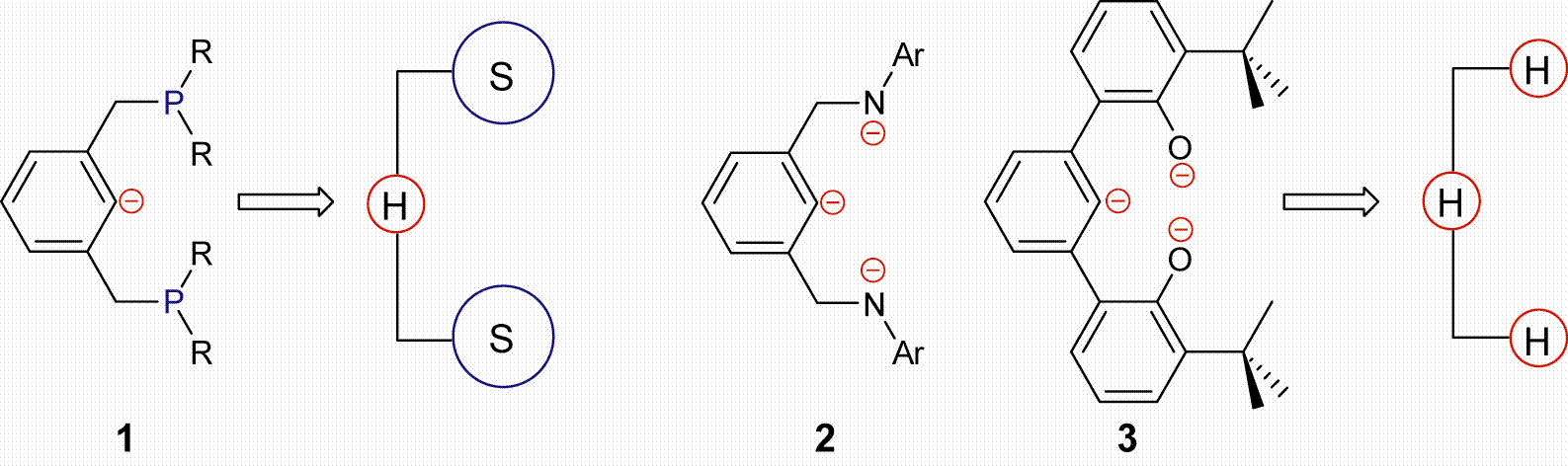
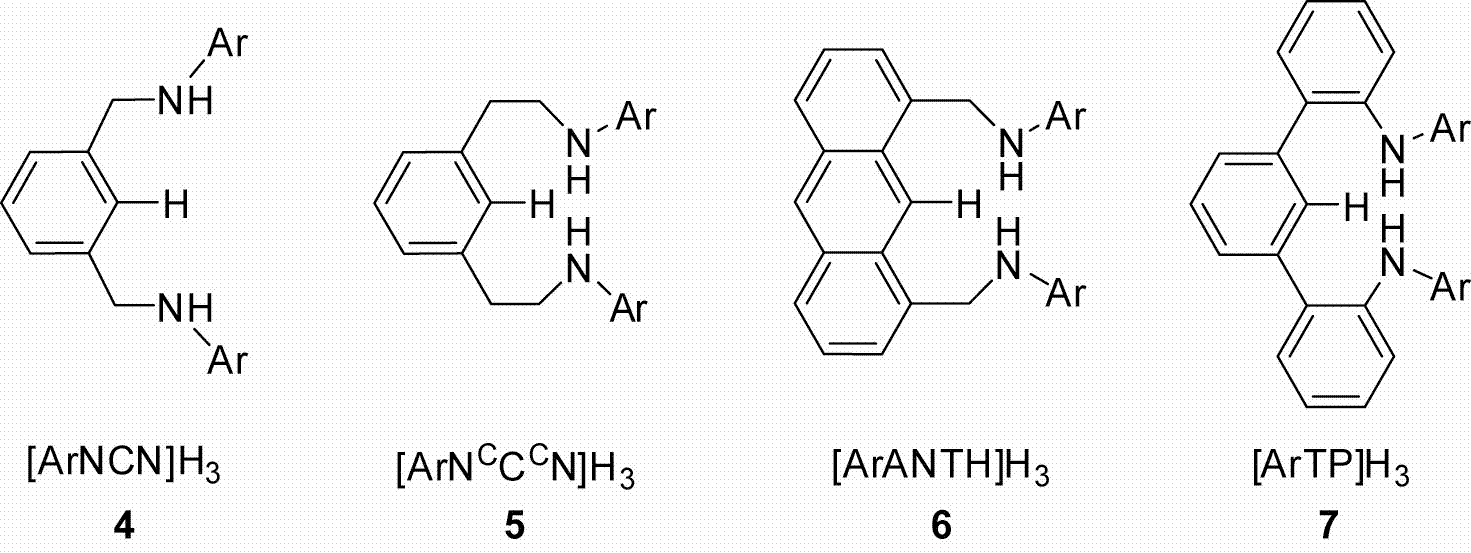
Chart 1.
One branch of our program centers on NCN3– pincer ligands and the following represents our progress to date. Most of the work was centered on the ligand type [ArNCN]H3 (where Ar = 2,6–iPrC6H3; 4–iPr) because these ligands are conveniently synthesized in high yield from inexpensive reagents (Chart 1). The pendant arms of the backbone were extended by one carbon and we were able to synthesize derivatives of the type [ArNCCCN]H3 (where Ar = Mes; 5–Mes, and 3,5–CF3C6H3; 5–CF3). To alter the rigidity of the backbone we successfully synthesized an anthracene derivative denoted as [ArANTH]H3 (where Ar = 2,6–iPrC6H3; 6–iPr).
Multiple approaches to metallation are conceivable and our first approach was to generate trilithio salts and then perform salt metathesis reactions with metal–halide substrates. Straightforward deprotonation with MeLi provided the trilithio salt derivative [2,6–iPrNCNLi3]2 (8) presented in Eq. 1. The molecular structure of 8 is presented in Figure 2. As a proof of concept these trianionic pincer ligands were shown to be capable ligands when bound to group IV metals. When MCl4 (M = Ti, Zr, Hf) are treated with ˝ equiv of 8, [2,6–iPrNCNMCl2]Li(solv)n derivatives are generated (M = Ti, 9–Ti; Zr, 9–Zr; Hf, 9–Hf). X-ray characterization of 9–Ti, 9–Zr, and 9–Hf was accomplished but only 9–Hf could be obtained in reproducible quantities. The molecular structure of 9–Hf is presented in Figure 2. The trianionic pincer ligand is bound in a terdentate fashion as planned which indicated our design was appropriate.
Various aryl groups can be installed easily but we discovered that judicious choice is necessary. For example, if the steric bulk of the aryl group is moved from the 2,6–position to the 3,5–position, the deprotonation chemistry is significantly altered. Treating [3,5–CH3C6H3]H3 (4–Me) with MeLi lead to deprotonation of the pendant arm methylene protons instead of the backbone ipso–CH. To solve this problem we synthesized the ipso–Br derivative [3,5–Me2ArNCN]H2Br (10) and subsequent treatment with n–BuLi provided the trilithio salt 11 cleanly and in excellent yield (Eq 7). Unfortunately, metallation with MCln salts again leads to SET and intractable mixtures. However, smooth metallation is achieved with HfCl4 but with the steric bulk now switched to the 3,5–position, two terdentate ligands bind to one Hf center to generate the bis-pincerate hafnium dianion [3,5–MeNCN]2Hf(Li2DME2) (12, Eq 7).
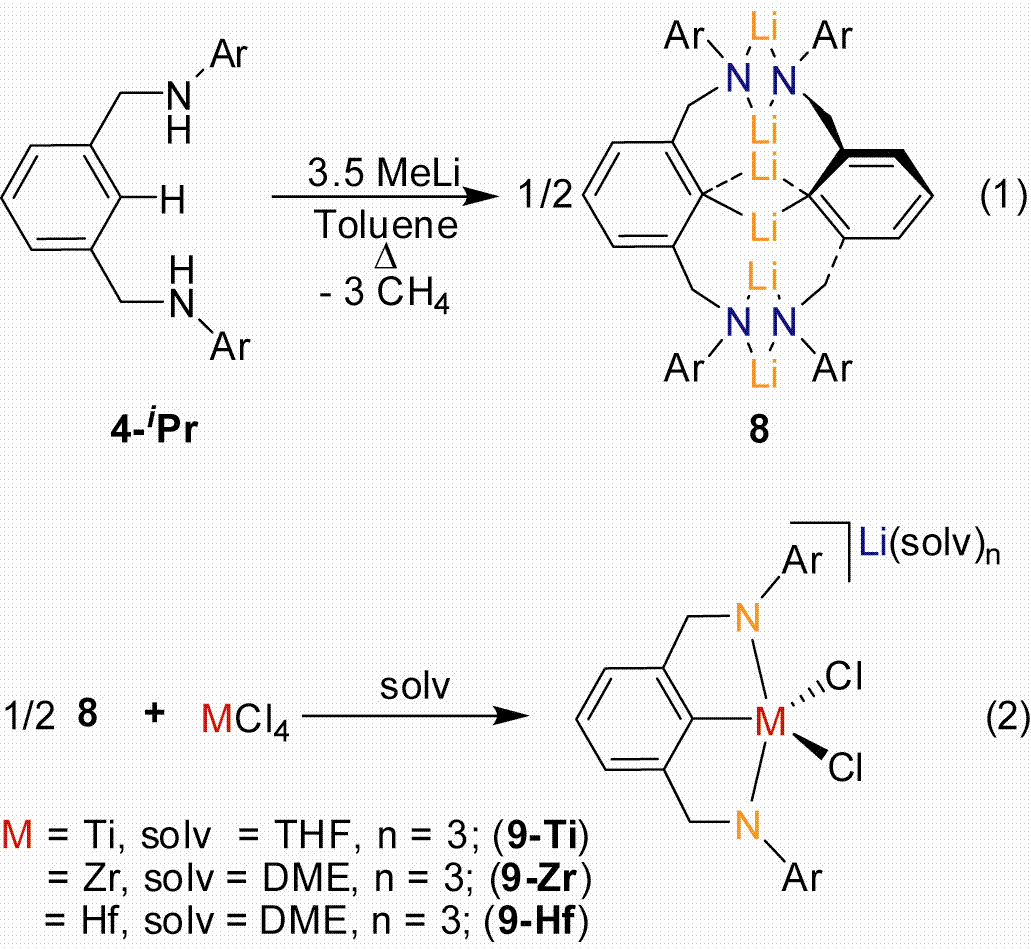
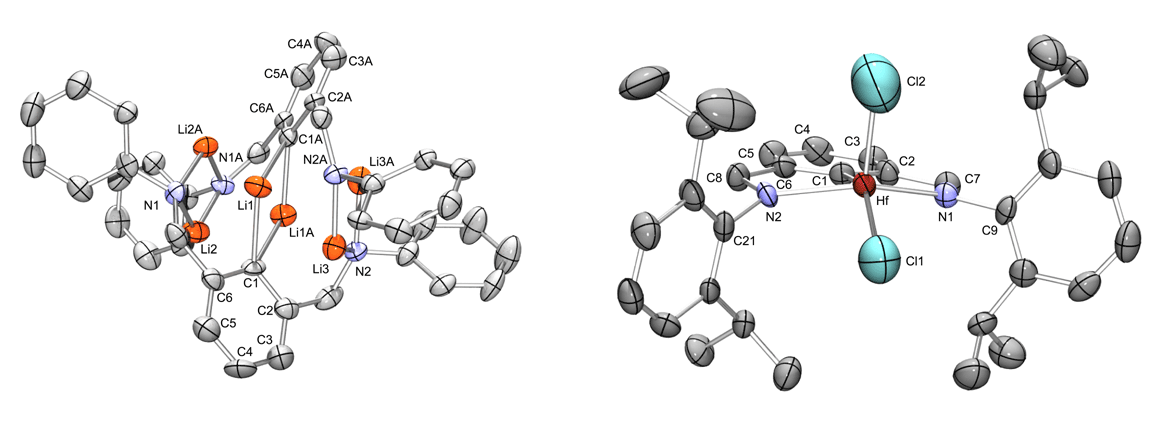
Figure 2. Molecular structures of 8 and 9–Hf with ellipsoids drawn at the 50% probability level and hydrogen atoms and IPr groups (8) removed for clarity.
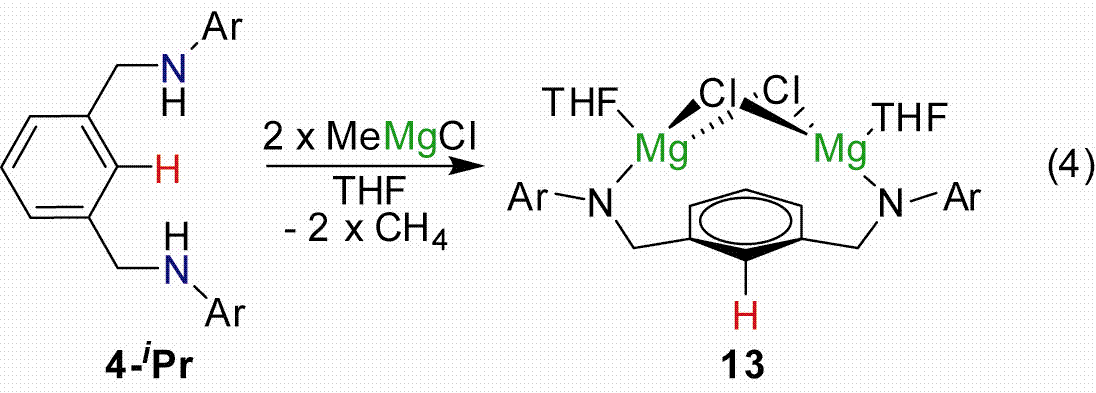
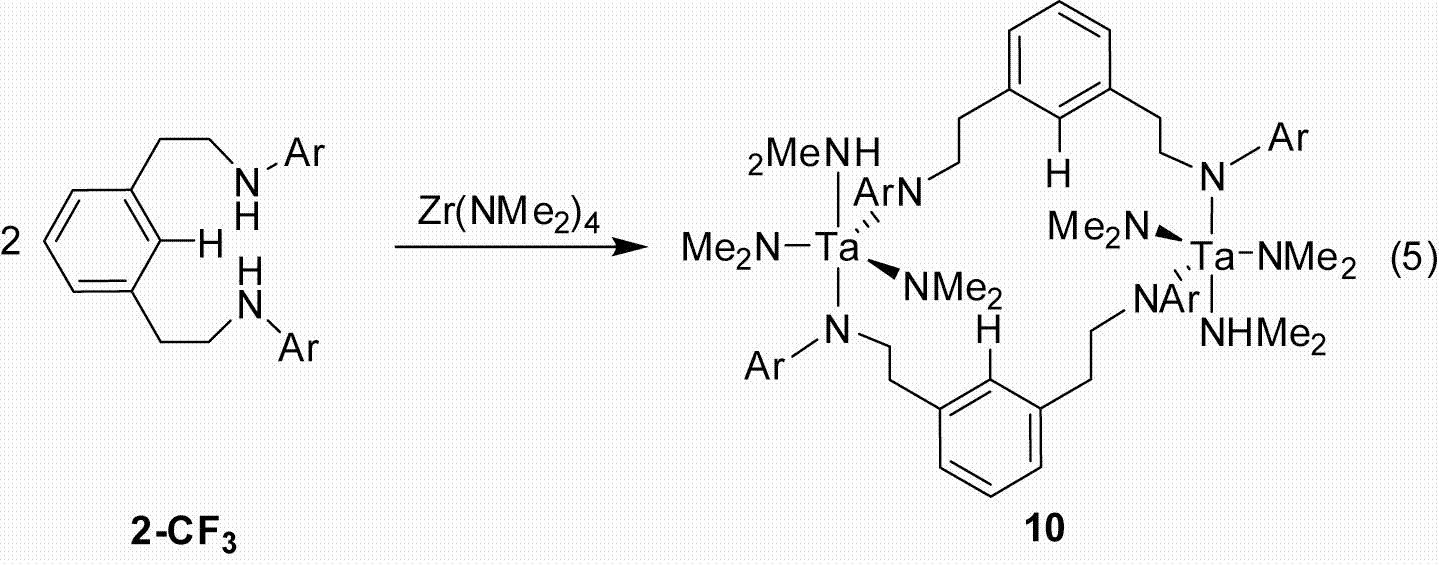
To minimize SET we synthesized the dimagnesium derivative [2,6–iPrNCN]HMg2(THF)2 (13, Eq. 8) but could not access a trianionic version (Figure 3). Consequently when metal substrates are introduced metallation does not occur, presumably because the aryl C–H bond is still intact. Alternatively starting with the neutral derivatives of either [2,6–iPrNCN]H3 (4–iPr) or [3,5–CF3ArNCCCN]H3 (5–CF3) lead to the formation of dimers. For example, treating Zr(NMe2)4 with 1 equiv of 5–CF3 provided the dimer {[3,5–CF3NCCCN]Zr(NMe2)2NHMe2}2 in quantitative yield (Eq. 9; Figure 4).
Figure 3. Molecular structure of 12 (left) and 13 (right).
Figure 4. Synthesis and molecular structure of 14 drawn at the 50% probability level.