
ACS PRF | ACS
All e-Annual Reports

44106-G6
Constrained Density Functional Theory as a Tool for Describing Electron Transfer
In addition to being among the most basic chemical processes known, electron transfer (ET) reactions also play an active role in natural and artificial photosynthesis, oxidative damage of DNA, respiration and single-molecule conductivity. In particular, we are interested in how long range ET is initiated and how the resulting electrochemical bias can be used to do work. In this case, one key factor is the reorganization energy - the energy the molecule gains by distorting to accommodate ET. One would like to be able to make quantitative predictions about the energy landscapes of these ET reactions using first principles calculations, such as density functional theory (DFT). Unfortunately, standard DFT fails qualitatively for long range ET because of the lack of particle-hole interactions in existing functionals. We have recently developed a method based on constrained density functional theory (CDFT) that is well-suited to describing long-range ET using diabatic states. Within this grant, we have proposed to build upon this basic realization to build a consistent framework for the description of electron transfer within DFT.
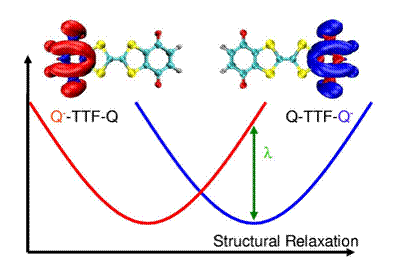
Figure 1 Our constrained DFT formalism gives us direct access to the reorganization energy (l), which determines the effective barrier migrating charges must overcome. For TTF-based molecules, like the diquinone show above, lowering the reorganization barrier could lead to novel low-dimensional molecular conductors.
In the first stage of this grant, we have implemented CDFT gradients, which allow for rapid molecular geometry optimization with a fixed amount of ET. At a fundamental level, this allows us to explore the diabatic potential energy surfaces involved in ET and also to address the magnitude and nature of inner sphere reorganization within the reaction. We have already used this method to predict the inner sphere reorganization in a few model donor acceptor systems, and the resulting methodology has given us significant insight into the spin-dependent rates of charge recombination in organic LEDs. A simple example of an application funded by this proposal is illustrated in Figure 1. Here, the fundamental parameter (inner sphere reorganization) is predicted quantitatively by our C-DFT calculations to be 13.3 kcal/mol – an extremely high value, even for an organic molecule. In turn, this value indicates that the quinine-TTF-quinone compound shown is does not function as a very good “molecular wire” as previously thought. Our simulations allow us to account for solvent effects through the use of established continuum solvation models and give us quantitative predictions about the energy landscape of ET in chemically relevant molecules.
In the second stage of this grant, we developed a means of computing the electronic coupling between the diabatic surfaces described above. This coupling plays a significant role in determining the rate of electron transfer and whether the reaction proceeds via an adiabatic or nonadiabatic pathway. Our initial applications of this technique showed that it gives accurate predictions of the coupling in several prototypical situations. By combining the gradients described above with this coupling prescription one obtains a unified ab initio picture of the kinetics of the electron transfer process.
Thanks to the successful leveraging of this grant by an NSF CAREER award, we have at this point accomplished all the major goals originally set out in our PRF proposal. Moving forward, we plan to extend this work by using the analytic gradients implemented within the PRF grant to perform ab initio molecular dynamics simulations of photoexcited ET. Here, we hope that our simulations will extend and deepen the understanding of molecular relaxation dynamics provided by ultrafast spectroscopic studies of ET excited states.