ACS PRF | ACS
All e-Annual Reports

41983-AC1
The Quest for Hydrocarbon Analogues of the Porphyrins
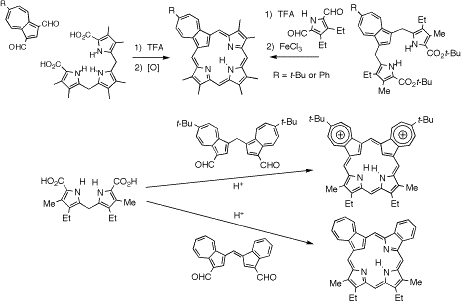
Azuliporphyrins have many intriguing properties but often have poor solubilities in organic solvents. In order to overcome this problem and to further study this system, a series of six azuliporphyrins with substituents on the seven-membered ring were prepared by two different ‘3 + 1' routes from 6-tert-butyl- and 6-phenylazulene. The substituted azulenes can be converted into dialdehydes under Vilsmeier-Haack conditions and these react with tripyrranes in the presence of TFA in CH2Cl2 to give azuliporphyrins in excellent yields. Alternatively, tripyrrane analogues can be prepared by reacting the substituted azulenes with an acetoxymethylpyrrole in the presence of acetic acid, and following a deprotection step these condensed with a pyrrole dialdehyde to give the related azuliporphyrins in 45-51% yield. Five of the azuliporphyrins were sufficiently soluble in CDCl3 to afford high quality proton and carbon-13 NMR data. The internal CH and NH resonances were observed near 3 ppm, although the precise values were dependent upon substituent effects. The presence of a tert-butyl group on the azulene moiety slightly enhanced the diatropicity of the macrocycle compared to the phenyl substituted azuliporphyrins. Polar solvents also increased the downfield shifts to the external protons by stabilizing the dipolar resonance contributors that are responsible for the carbaporphyrinoid aromatic character. A tert-butyl substituted azuliporphyrin also gave X-ray quality crystals and this allowed the first structural analysis of a free base azuliporphyrin to be conducted. The macrocycle is near planar and the azulene unit was only tilted out of the plane by 7.4o. An analysis of the bond lengths suggests that a 17 atom delocalization pathway significantly contributes to the aromatic properties of this system. Protonation of azuliporphyrins affords dications with enhanced diamagnetic ring currents where the internal CH shifts to ca. -3 ppm. Again, the chemical shifts are influenced by the substituents and the presence of an electron-donating tert-butyl group on the azulene subunit increases the macrocyclic diatropicity. Two of the substituted azuliporphyrins were reacted with nickel(II) acetate or palladium(II) acetate in DMF to give the corresponding organometallic derivatives, and these stable complexes were isolated in excellent yields. Addition of pyrrolidine to NMR solutions of 23-substituted azuliporphyrins demonstrated that nucleophilic addition products were present in equilibrium with the parent porphyrinoids but these adducts are less favored than for azuliporphyrins lacking the 23-substituents. Although nucleophilic attack of a peroxide anion is believed to be the first step in the conversion of azuliporphyrins to benzocarbaporphyrins with t-BuOOH and KOH, the tert-butyl or phenyl substituents in azuliporphyrins and did not inhibit this chemistry. Two benzocarbaporphyrin products were isolated and characterized in each case, and mechanisms are proposed to explain the origins of these oxidative ring contraction products. Azulene has also been incorporated into adj-dicarbaporphyrinoid systems with borderline aromatic characteristics. For these syntheses, ‘2 + 2' MacDonald condensations were used to condense a dipyrrylmethane with a fulvene dialdehyde or a diformyl diazulenylmethane. These fully conjugated porphyrinoids are formed in good yields and unlike previously synthesized opp-dicarbaporphyrins they are stable compounds that will allow further investigations into their reactivity (e.g., towards metalation).