
ACS PRF | ACS
All e-Annual Reports

43281-AC7
Molecular Organization in Phospholipid Bilayers Containing Trans Fatty Acids Studied by Solid State 2H NMR
The objective of the project is to employ solid state 2H NMR, complemented by MD simulations, to elucidate molecular organization in a phospholipid bilayer containing a trans fatty acid (TFA). This report focuses on the synthesis of sn-1 substituted phosphatidylcholines (PC) bearing deuterated anlogs of elaidic (t18:1) and, for comparison, oleic (c18:1) acid.
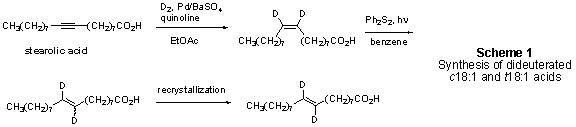
Synthesis of Dideuterated Acids
[9,10-D2]c18:1 acid was prepared by selective deuteration of stearolic acid, followed by photochemical isomerization using a pyrex-filtered light source to produce [9,10-D2]t18:1 acid with >80% yield (Scheme 1). Cis and trans isomers were separated by repeated crystallization from ethanol/water.
Synthesis of Perdeuterated Acids
The corresponding perdeutuerated acids were constructed by acetylide coupling of two distinct C8 precursors, to produce D31-stearolic acid as key intermediate. Synthesis of the C1-C8 portion started with D14-suberic acid, which was reduced and desymmetrized to yield the corresponding D16-8-bromo-octanol (Scheme 2). Although literature suggested that reduction of suberic acid was easily accomplished with borane-THF, we found that the use of LiAlD4 in THF at rt for 12h produced better results. As the diol is soluble in water, we chose to avoid an extractive work-up and instead performed silica gel chromatography on the reaction mixture after precipitation of the bulk of aluminum oxides by the careful addition of small amounts of NaOH (aq) and methanol. The diol was subsequently reacted with 1.4 equiv HBr in toluene and the resulting bromoalcohol converted to the corresponding THP-ether.

Reaction of lithium acetylide-EDA to D17-1-bromooctane in DMSO produced the C10 acetylene, which was subsequently coupled to the above THP-protected bromide (Scheme 3). This reaction was examined under a variety of condition and although it is common to prepare the more reactive iodide electrophile, we found that this procedure caused a significant increase in decomposition products. Ultimately, the coupling reaction was best accomplished by refluxing the acetylide anion (deprotonation with 1 equiv Bu- or MeLi) for 2 days with the THP-protected bromide in the presence of a catalytic amount of NaI.
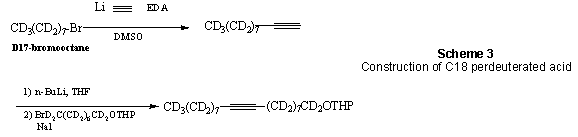
Once the carbon chain had been assembled, the THP ether was removed and the corresponding alcohol was oxidized to produce D31-stearolic acid. Oxidation of acetylenic alcohols is typically performed with Jones reagent or PDC; however, because this reaction is usually inefficient, these and alternate methods were more closely investigated. Consistent with the literature, Jones oxidation was a low yielding reaction and in addition, we discovered that it was difficult to quantitatively recover the product from the aqueous layer. Ultimately, we chose to perform sequential oxidations i.e., PCC oxidation to the aldehyde, followed by sodium chlorite (1 equiv) oxidation of the crude aldehyde in a rapidly stirring biphasic mixture of ether and buffered water. Despite our precautions, the second oxidation was low yielding, and starting from 1 g of D14-suberic acid, only 250 mg of D31-stearolic acid was obtained. Deuteration of this material went smoothly to produce D33-oleic acid, which will be used for planned PC derivatization.
Synthesis of PC
The final steps of the PC synthesis involve the coupling of oleic (or elaidic) acid to the sn-1 position of glycerol-3-phosphatidylcholine (GPC). Although this reaction would appear straightforward, several important issues must be considered including i) the solubility of the GPC, ii) the equivalency and form of carbonyl electrophile, and iii) the purification method used for the lyso-PC that forms. GPC (including the corresponding cadmium salt) is poorly soluble in all solvents typically used for (non-acidic) ester couplings and although the primary hydroxyl of GPC is more reactive, the secondary hydroxyl will also react due to its higher effective concentration in solution as sn-1-acyl-2-hydroxy-PC forms. To circumvent these problems and others, excess fatty acid anhydride (>4 equiv) is typically reacted with a heterogenous mixture of mostly insoluble GPC in CHCl3, with dimethylamino pyridine as the superior nucleophilic catalyst. The resulting diacyl-PC is converted to the lyso structure by enzymatic hydrolysis with phospholipase (PLA2). Even under the best conditions, we found this reaction to be poor yielding and wasteful with respect to fatty acid required.
Recently we have explored a variety of alternate coupling methods using (protonated) oleic and stearic acid as model fatty acids. Currently, optimal coupling is achieved by DCC mediated coupling in DMF as solvent, using DMAP as catalyst. Purification is performed by sequential C18 and silica gel chromatography (elution of lyso-PC from SiO2 with 40/50/10 CHCl3/MeOH/0.25M aq acetic acid). Under these reaction conditions, we observe high selectivity for lyso formation, although the reaction yield is only modest (between 40-50%). Further optimization is planned before this reaction is applied to the preparation of the requisite deuterated PC's.