
ACS PRF | ACS
All e-Annual Reports

45592-AC5
Investigation of Catalysis by Nanoparticles and Metal-Ion Complexes Embedded in Multilayer Polyelectrolyte Films
Development of effective heterogeneous catalysts is vital for creating environmentally benign, energy efficient reactions. Although such catalysts may be relatively expensive, they can be readily recycled. This research aims at synthesizing selective, heterogeneous catalysts through immobilization of metal nanoparticles in multilayer polyelectrolyte films both on alumina beads and in membranes. Nanoparticles are attractive catalysts because of their high surface area and tunable electronic properties, and embedding these materials in polyelectrolyte films both prevents particle aggregation and restricts access to catalytic sites to impart remarkably high selectivities.
A commercial catalyst, 5% Pd on alumina, showed hydrogenation selectivities <2.2 for all of the compounds in Table 1, suggesting that differences in rates of hydrogenation using [PAA/PEI-Pd(0)]3PAA films on alumina (Table 1) are not due primarily to electronic factors. Thus, the mechanism behind the selectivity of the [PAA/PEI-Pd(0)]3PAA catalysts appears to be hindered access of multiply substituted double bonds to catalytic sites, and we are currently investigating how this selectivity may vary with particle size. In a The funding from this grant was vital in starting this work and supporting two graduate students, David Dotzauer and 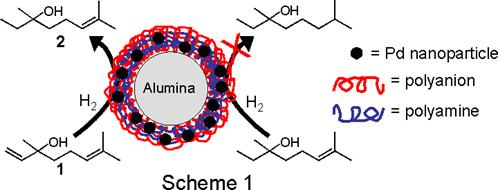 With funding from this grant, we demonstrated that Pd nanoparticles embedded in polyelectrolyte films show remarkable intramolecular selectivity in the hydrogenation of alkenes, with discrimination being based on the degree of substitution of the double bond. These catalysts are prepared using layer by layer deposition of poly(acrylic acid) (PAA) and a polyethyleneimine-Pd2+ (PEI-Pd2+) complex on alumina particles. Subsequent reduction of Pd2+ to Pd(0) with NaBH4 yields the nanoparticles embedded in the polyelectrolyte film, and TEM images of such films deposited on carbon-coated Cu grids show nanoparticle diameters of 1-3 nm. Scheme 1 represents a particularly successful example of selective catalysis where Pd nanoparticles embedded in PAA/PEI films on alumina catalyzed hydrogenation of 3,7-dimethyl-1,6-octadien-3-ol, 1, to 3,7-dimethyl-6-octen-3-ol, 2, with no detectable doubly reduced product.
With funding from this grant, we demonstrated that Pd nanoparticles embedded in polyelectrolyte films show remarkable intramolecular selectivity in the hydrogenation of alkenes, with discrimination being based on the degree of substitution of the double bond. These catalysts are prepared using layer by layer deposition of poly(acrylic acid) (PAA) and a polyethyleneimine-Pd2+ (PEI-Pd2+) complex on alumina particles. Subsequent reduction of Pd2+ to Pd(0) with NaBH4 yields the nanoparticles embedded in the polyelectrolyte film, and TEM images of such films deposited on carbon-coated Cu grids show nanoparticle diameters of 1-3 nm. Scheme 1 represents a particularly successful example of selective catalysis where Pd nanoparticles embedded in PAA/PEI films on alumina catalyzed hydrogenation of 3,7-dimethyl-1,6-octadien-3-ol, 1, to 3,7-dimethyl-6-octen-3-ol, 2, with no detectable doubly reduced product. 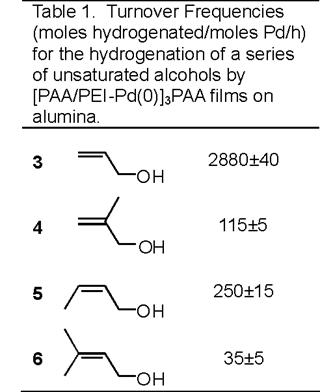 Pd nanoparticles embedded in PAA/PEI films also exhibit high intermolecular selectivities as shown in Table 1. The selectivities (ratios of turnover frequencies) for hydrogenation of the monosubstituted double bond in 3 relative to hydrogenation of the disubstituted double bonds in 4 and 5 are 25 and 12, respectively. For comparison, the prototypical homogeneous catalyst for such reactions, tris(triphenylphosphine)rhodium(I) chloride (Wilkinson's catalyst), shows a rate of hydrogenation of 1-hexene in benzene that is only 1.1 times higher than that for 2-methylpent-1-ene. Additionally, the selectivity for nanoparticle-catalyzed hydrogenation of 3 relative to 6 is 82, which is comparable to the selectivity of Wilkinson's catalyst. Importantly, the nanoparticle/alumina system is a heterogeneous catalyst that can be easily separated from the reaction mixture.
Pd nanoparticles embedded in PAA/PEI films also exhibit high intermolecular selectivities as shown in Table 1. The selectivities (ratios of turnover frequencies) for hydrogenation of the monosubstituted double bond in 3 relative to hydrogenation of the disubstituted double bonds in 4 and 5 are 25 and 12, respectively. For comparison, the prototypical homogeneous catalyst for such reactions, tris(triphenylphosphine)rhodium(I) chloride (Wilkinson's catalyst), shows a rate of hydrogenation of 1-hexene in benzene that is only 1.1 times higher than that for 2-methylpent-1-ene. Additionally, the selectivity for nanoparticle-catalyzed hydrogenation of 3 relative to 6 is 82, which is comparable to the selectivity of Wilkinson's catalyst. Importantly, the nanoparticle/alumina system is a heterogeneous catalyst that can be easily separated from the reaction mixture.  second reduction reaction, we found that nanoparticles are also capable of selectively reducing nitro functionalities in the presence of other reducible groups such as chlorine and nitriles. Such selective reduction is important for the production of functionalized anilines that can serve as precursors for fine chemical synthesis. In these experiments, we used NaBH4 as a reducing agent and nanoparticles immobilized in porous alumina membranes as catalysts (Scheme 2). (H2 can also be employed as the reducing agent.) The use of membranes avoids the need to collect the catalyst and also allows fine control over the residence time in the presence of the catalyst, which should in principle afford control over the degree of reaction. As a proof of concept experiment, we attempted to reduce 2-nitrotoluene to 2-nitrosotoluene rather than 2-aminotoluene by increasing the linear velocity of the reaction solution through the membrane. At a linear velocity of 0.05 cm/sec (residence time of 120 msec), the product of the reaction was 71% 2-aminotoluene and 29% 2-nitrosotoluene with no residual 2-nitrotoluene. In contrast, at a linear velocity of 0.8 cm/sec (residence time of 12 msec), the reaction product was 37% 2-aminotoluene and 63% 2-nitrosotoluene. Future studies aim at better exploiting control of residence time to control product distribution.
second reduction reaction, we found that nanoparticles are also capable of selectively reducing nitro functionalities in the presence of other reducible groups such as chlorine and nitriles. Such selective reduction is important for the production of functionalized anilines that can serve as precursors for fine chemical synthesis. In these experiments, we used NaBH4 as a reducing agent and nanoparticles immobilized in porous alumina membranes as catalysts (Scheme 2). (H2 can also be employed as the reducing agent.) The use of membranes avoids the need to collect the catalyst and also allows fine control over the residence time in the presence of the catalyst, which should in principle afford control over the degree of reaction. As a proof of concept experiment, we attempted to reduce 2-nitrotoluene to 2-nitrosotoluene rather than 2-aminotoluene by increasing the linear velocity of the reaction solution through the membrane. At a linear velocity of 0.05 cm/sec (residence time of 120 msec), the product of the reaction was 71% 2-aminotoluene and 29% 2-nitrosotoluene with no residual 2-nitrotoluene. In contrast, at a linear velocity of 0.8 cm/sec (residence time of 12 msec), the reaction product was 37% 2-aminotoluene and 63% 2-nitrosotoluene. Future studies aim at better exploiting control of residence time to control product distribution.