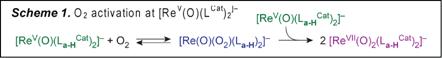ACS PRF | ACS
All e-Annual Reports

45130-G3
Redox Coupling for Multielectron Small-Molecule Activation
A. Overview and Significance
Solutions to outstanding problems in laboratory synthesis, fine- and commodity-chemicals production, and generation of chemical fuels for the energy challenge will hinge on molecular catalysts with a multielectron capacity. In this context, the use of oxygen as a terminal oxidant for selective catalytic transformations of small-molecule organic substrates is extremely desirable. Multielectron aerobic oxidation reactions that mimic biological oxygenases and incorporate one or both oxygen atoms from O2 into the organic substrate, or oxidases which couple substrate oxidation to reduction of dioxygen have seen recent attention. Traditional inorganic and organometallic approaches to such aerobic oxidations employ transition metal ions with redox-inert ancillary ligands. By comparison, the ability of redox-active ligands to couple small-molecule redox transformations with O2 reduction has been largely unexplored. Presented here are highlights of new oxidase and oxygenase-type reactions from our lab, where a redox-active ligand is an essential component of the novel catalytic reactivity.
B. Progress Report
B.1 Intramolecular Proton-Coupled Electron Transfer (PCET) for Oxidase Reactivity. In this research focus, substrate oxidation is coupled to O2 reduction through a cascade of ligand-mediated intramolecular PCET reactions. Our approach derives specificity from pre-assembly of a catalyst-substrate complex prior to the PCET event.
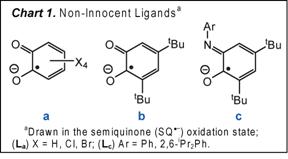
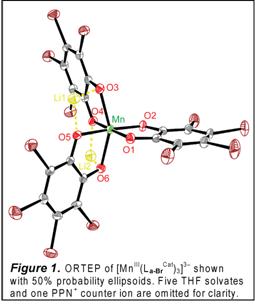
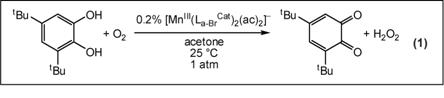
We have prepared new five- and six-coordinate complexes based on the square-planar [MnIII(La-BrCat)2]– core (La-Br, X = Br; Chart 1). The propensity of [MnIII(La-BrCat)2]– to bind both alcohol as [MnIII(La-BrCat)2(MeOH)]– and ketone as [MnIII(La-BrCat)2(Me2C=O)2]–, and the ability of related [MnIII(La-BrCat)2(L)n]– materials to effect reduction of O2 to H2O2, led us to believe that [MnIII(La-BrCat)2(MeOH)]– may be active in aerobic oxidative dehydrogenation reactions. Yet contrary to earlier reports, [MnIII(La-BrCat)2(MeOH)]–, [MnIII(La-BrCat)2(Me2C=O)2]– and [MnIII(La-BrCat)2(OPPh3)]– are indefinitely stable in solution under air. Instead, [MnIII(La-BrCat)3]3– (Figure 1) is the air-sensitive material. Addition of tetrabromo-o-catecholate to [MnIII(La-BrCat)2(Me2C=O)2]– gives [MnIII(La-BrCat)3]3–, which reacts with O2 to generate [MnIV(La-BrCat)3]2– in minutes. Blue [MnIV(La-BrCat)3]2– is itself unstable towards O2, slowly converting to [MnIII(La-BrCat)2(Me2C=O)2]– and tetrabromo-o-quinone. The overall reaction is an aerobic dehydrogenation reaction which converts tetrabromo-o-catechol to tetrabromo-o-quinone. We postulated that a more easily oxidized catechol substrate could effect rapid catalytic turnover. With 0.2% catalyst loading of [MnIII(La-BrCat)2(ac)2]– in acetone, quantitative conversion of 3,5-di-tert-butylcatechol to 3,5-di-tert-butylquinone is achieved in ca. 300 minutes under 1 atm O2 and at ambient temperature (eq 1). This preliminary success in catalyst design is paving the way for extensions to other aerobic hydrogenation reactions that proceed by intramolecular PCET.
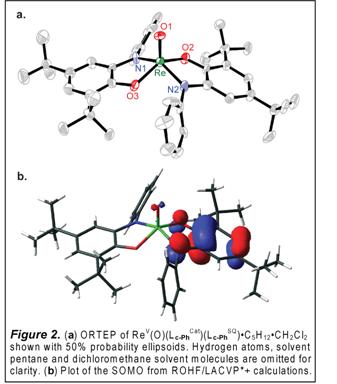
B.2 Intramolecular Redox Coupling for Oxygenase Reactivity. This research aims to elaborate a novel, odd-electron reactivity at terminal metal oxo ligands by coupling to redox-active quinoid cofactors. During the last year we have demonstrated that catecholate ligands can act as electron reservoirs to transfer electrons to and from closed-shell oxos, allowing even-electron reactions to occur by a series of single-electron steps. We have synthesized and completely characterized a library of five- and six-coordinate complexes of the general formula [ReV(O)(LCat)2(L)x]– (LCat = La-Lc; L = PPh3, OPPh3; x = 0, 1). One e– oxidiation of [ReV(O)(Lc-PhCat)2]– gives as ReV(O)(Lc-PhCat)(Lc-PhSQ) with one o-amidophenolate chelate and one o-iminosemiquinone radical ligand (Figure 2). Exposure of CH3CN solutions of olive green [ReV(O)(LCat)2]– to 1 atm O2 gives clean and quantitative conversion to purple [ReVII(O)2(LCat)2]–. In dilute MeOH solutions, reaction of [ReV(O)(La-HCat)2]– with 1 atm O2 first affords a blue intermediate before slowly converting to [ReVII(O)2(La-HCat)2]–. Removal of O2 from the blue solutions cleanly regenerates [ReV(O)(La-HCat)2]–, while concentration gives rapid conversion to [ReVII(O)2(La-HCat)2]–. The blue complex is an O2-adduct [Re(O)(O2)(La-H)2]–. The overall reaction requires two equivalents of ReV, consistent with the stoichiometry indicated in Scheme 1. The redox-active ligand environment afforded by the catecholate chelates is a key feature of this unique reactivity. Parallels can be drawn to first-row transition metal complexes that activate O2 by a two-electron process (Mn+ + ˝O2 → M(n+2)+=O) yet proceed through a series of single-electron steps with symmetric M(n+1)+–O2–M(n+1)+ peroxo-bridged intermediates. Here the catecholate functionality in [ReV(O)(LCat)2]– provides an internal redox buffer to stabilize a symmetric transition state for O=O bond homolysis, effectively lowering the barrier to the net two-electron process by allowing it proceed through a series of ligand-mediated one-electron redox steps. Addition of PAr3 to [ReVII(O)2(LCat)2]– effects rapid, quantitative conversion to [ReV(O)(LCat)2]– and the corresponding phosphine oxide. Coupling of this O-atom transfer process to O2 activation by [ReV(O)(LCat)2]– completes a catalytic cycle for aerobic phosphine oxidation. Experiments are currently in progress to assay the scope of this reactivity with an eye toward developing other oxygenase-type reactions derived from O=O bond homolysis by redox-active ligand complexes of 4d and 5d metal ions. C. Career Impact The work supported by this grant is the subject of two manuscripts in preparation. PRF support has additionally been acknowledged at presentations made at the Inorganic Gordon Research Conference.