ACS PRF | ACS
All e-Annual Reports

43207-AC10
Synthetic and Theoretical Studies of Electron Transport in Macromolecules and Nanoassemblies
Our research effort is a combined synthetic and molecular simulation approach to using ferritin as a molecular “synthetic machine” to self-assemble novel nanoparticles in the core of the ferritin assembly. To date, we have developed several synthetic strategies to incorporate novel nanocrystalline materials inside the ferritin cage, and over the last year we have successfully modeled the electrostatic properties of the ferritin cage and we are currently using this information to extract dynamics data of ion mobility through the 3-fold channels in the ferritin structure using molecular simulations.
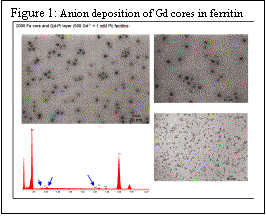 The discovery in the Watt lab that anions can be co-deposited in ferritin using the common iron deposition process of incubating ferritin with Fe2+ ion in the presence of oxygen and the appropriate anion opened new synthetic possibilities for preparing nanomaterials in ferritin. We have examined the process of iron and anion deposition into ferritin by examining halides, and trigonal planar anions to determine their suitability for deposition. Figure 1 shows the effectiveness of anion deposition of Gd cores.
The discovery in the Watt lab that anions can be co-deposited in ferritin using the common iron deposition process of incubating ferritin with Fe2+ ion in the presence of oxygen and the appropriate anion opened new synthetic possibilities for preparing nanomaterials in ferritin. We have examined the process of iron and anion deposition into ferritin by examining halides, and trigonal planar anions to determine their suitability for deposition. Figure 1 shows the effectiveness of anion deposition of Gd cores.
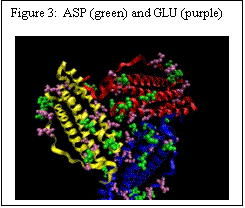
 Ferritin has a cage like structure, made up of 24 equivalent sub-units. As shown in Figure 2, at the confluence of these subunits, there are 3-fold and 4-fold channels – the former allow ions to move between the exterior of the cage and the core, and the latter are primarily electron transfer conduits. The 3-fold channels are lined with ASP and GLU amino acids (Figure 3).
Ferritin has a cage like structure, made up of 24 equivalent sub-units. As shown in Figure 2, at the confluence of these subunits, there are 3-fold and 4-fold channels – the former allow ions to move between the exterior of the cage and the core, and the latter are primarily electron transfer conduits. The 3-fold channels are lined with ASP and GLU amino acids (Figure 3).
A number of reducing agents have been found to effect reduction of the Fe(III) mineral core. The mode of entry of reductants into ferritin has not been established but the polarity of the three-fold pore sites indicates that polar reducing agents might enter that way. To address the issue of ion transport in ferritins, we focus on the following questions:
· Do reducing agents bind to the protein surface and where?
· Can molecular dynamics simulations and essential dynamics sampling determine if the pores expand and contract sufficiently during large scale breathing motions to allow mid-sized reducing agents through.
We have used the AutoDock suite of programs and the Lamarckian genetic search algorithm in conjunction a semi-empirical free energy forcefield with charge-based desolvation to determine the best docking sites. Our docking studies have used the human H chain ferritin crystal structure and the agents reduced riboflavin (RR) and flavin mononucleotide (FMN).
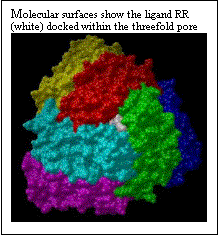

The polar riboflavin (RR) binds primarily within the threefold pore and one additional site at the junction of two ferritin subunits, as shown in Fig. 4. Only six subunits around the pore are displayed. Six acidic residues (ASP+GLU) line the threefold pore. In Figure 5, the charged flavin mononucleotide is seen to bind primarily at the junction of two ferritin subunits. This area has the polar and basic residues ASN and LYS. The binding pocket, though shallow is clearly visible in Fig. 5. Binding energies for the minimum energy docked conformations at the different sites along with the inhibition constants are given in Table 1.
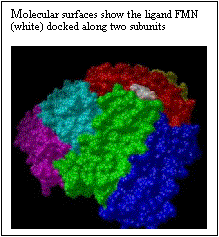
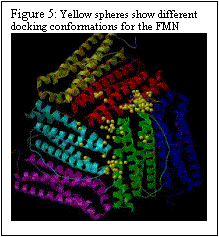
Table I: The binding energies (kcal/M) and inhibition constants, Ki (mM)
site binding energy | inhibition constant | |
RR threefold site | 8.01 | 1.34 |
FMN (subunit junction) | 7.89 | 1.42 |
RR four-fold site | 6.92 | 8.53 |
FMN fourfold site | 5.45 | 100.7 |
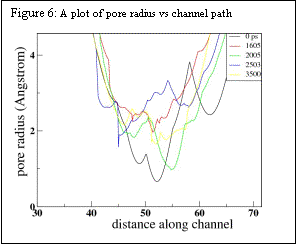
Constant temperature molecular dynamics simulations at 310.15 K using the Berendsen Thermostat were performed using GROMACS simulation package. The system was simulated for 50 ps by applying position restraints on the protein and letting the solvent equilibrate. Position restraints were removed and the simulation was then run at constant temperature and pressure for 3.5 ns. The pore radius at approximate intervals of 200 ps was determined using the program HOLE. Significant distortion of the pore as well as change in pore size occurs in 3.5ns. As shown in Fig. 6, constriction in the pore changes in size by about 2Ǻ on this time scale – significant, but not enough to allow large molecules to enter on this short nanosecond time scale.
Unfortunately, large scale protein motions occur on longer time-scales which may range from nano- to micro-seconds, and conventional molecular dynamics trajectories span at most to the nanosecond range. One of the methods to reduce the dimensionality of the problem is to isolate motion along only a few collective coordinates which would adequately represent the over-all large scale motion of the system. Future work that uses essential dynamics analysis or covariance analysis to accomplish this using 20-40 ns MD simulation trajectories is currently underway.