ACS PRF | ACS
All e-Annual Reports

45401-AC3
Titanium-Mediated and -Catalyzed Amine Allylation: Applications, Mechanism, and Implications for '[3,3]-Sigmatropic' Rearrangements Involving Metal-Ligand Multiple Bonds
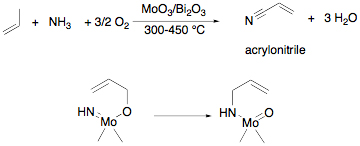
Allyl transfer to a metal ligand multiple bond substituent is a key step in many catalyses. This reaction is generally assumed to occur through [3,3]-sigmatropic rearrangement similar to a Cope or Claisen rearrangment. For example, the SOHIO process[1] for propylene ammoxidation is believed to involve the transfer of an allyl group to a molybdenum imido on the way to acrylonitrile production (Scheme 1). One proposal for the key C–N bond formation is sigmatropic migration of an allyl in this important industrial process. Alkoxide [3,3]-sigmatropic rearrangements are also drawn in the mechanism for allyl alcohol isomerization reactions catalyzed by transition metal oxides like OV(OPri)3. These reactions are used, for example, in isomerization of terpenes like linolool in the fragrance industry.[2]
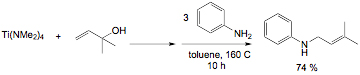
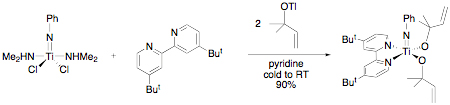
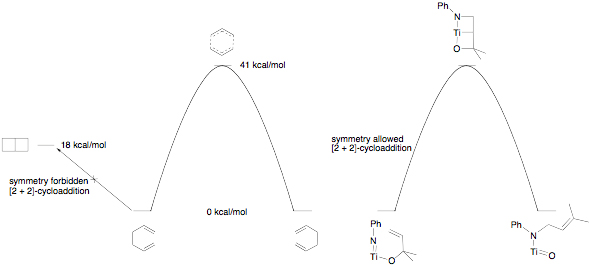
Scheme 1. Propylene ammoxidation (top) and the proposed [3,3]-sigmatropic rearrangement in the heterogeneous catalyst that forms the new C–N bond (bottom). In our research, we have developed a new methodology for the synthesis of secondary allyl amines based on a titanium-mediated process.[3] In these reactions, an alcohol is added to commercially available Ti(NMe2)4 followed by amine. The reaction is believed to involve in situ generation of a titanium imido alkoxide. The allyl group transfers from oxygen to nitrogen selectively generating the secondary amine with allylic transposition (Scheme 2). In this chemistry, the rearrangement was inhibited by placing a methyl group in the 2-position of the allyl alcohol, which should not effect a [3,3]-sigmatropic rearrangement (Scheme 1, bottom). An alternative mechanism is a [2 + 2]/retro-[2 + 2] cycloaddition pathway involving two fused four-membered metallacycles. Scheme 2. Example of the secondary allylic amine synthesis mediated by Ti(NMe2)4. In the last year, we have developed a method for the synthesis of an unknown class of titanium imido allyl alkoxide complexes to test this mechanism. The synthesis for the 1,2-dimethylallylalkoxide is shown in Scheme 3. The sequence begins with a titanium imido complex available using a route developed by Lorder and coworkers.[4] A 1,1,2-trimethylallylalkoxide derivative was prepared similarly. Scheme 3. Synthesis of titanium imido allyl alkoxides. The derivative without the 2-methyl group rearranged to produce the expected secondary amine after hydrolysis with a rate constant about 100 times smaller than the complex without the 2-methyl group, suggesting a similar mechanism to the Ti(NMe2)4 in situ generated system (Scheme 2). In this isolable imido system, we are measuring a secondary kinetic isotope effect (SKIE) in the allyl transfer in the case where the proton is replaced with a deuterium in the 2-position. Consistent with a [2 + 2]/retro-[2 + 2]-cycloaddition pathway, the preliminary experimental values for the rate constants suggest a large inverse isotope effect at that carbon, consistent with rehybridization in the transition state. The mechanistic studies completed to date are consistent with our proposed pathway for allyl transfer from the allylic alkoxide to imido nitrogen involving a bicyclic intermediate or transition state. Our current understanding of the mechanistic situation in comparison with the well-known Cope [3,3]-sigmatropic rearrangement[5] is shown in Figure 1. The organic system has a potential low-energy intermediate in bicyclo[2.2.0]hexane; however this intermediate is kinetically inaccessible due to symmetry restrictions on thermal allowability of organic [2 + 2]-cycloadditions. In the related titanium system, the [2 + 2]-cycloaddition product is symmetry allowed and accessible as either an intermediate or transition state. DFT calculations on a model system, Ti(NPh)[OC(Me)2CHCH2](NH2)(NH3), using several different functionals suggest the bicyclic complex is an intermediate. While completing these SKIE studies on the allyl transfer, we are beginning studies on the enantioselectivity of the allyl transformation and the concurrently discovered alkyl migration reaction. It is hoped that as these mechanistic studies proceed new reaction pathways will be discovered and methodologies for optimizing those pathways will be found, which will act as inspiration for us and others to develop efficient new bond forming reactions. [1]. Grasselli, R. K. Catalysis Today 2005, 99, 23. [2]. Parshall, G. W.; Ittel, S. D. Homogeneous Catalysis, 2nd Ed.; Wiley Interscience: New York, 1992. [3]. Ramanathan, B.; Odom, A. L. J. Am. Chem. Soc. 2006, 128, 9344. [4]. Lorber, C.; Choukroun, R.; Vendier, L. Eur. J. Inorg. Chem. 2006, 4503.