
ACS PRF | ACS
All e-Annual Reports

43444-AC4
Development of Photoreactive Precursors for Time and Spatially Resolved Generation and Direct Study of p-Benzyne Diradicals
The goal of this project is to develop methods for the efficient photochemical generation of the strained cyclic enediyne compounds, which undergo a very rapid thermal Bergman cycloaromatization. The photo-induced formation of highly reactive enediynes opens the opportunity for the spatial and temporal control of the p-benzyne diradical generation and for the direct spectroscopic investigation of this reactive intermediate. We also studied the reversibility of the Bergman cyclization, as well as the feasibility of controlling the enediyne reactivity by changing the position of the keto-enol equilibrium.
To achieve these goals, we have synthesized eleven (1a) and twelve (1b) – membered ring cyclic enediyne compounds incorporating 2-diazo-1,3-diketo functionality (Scheme 1).
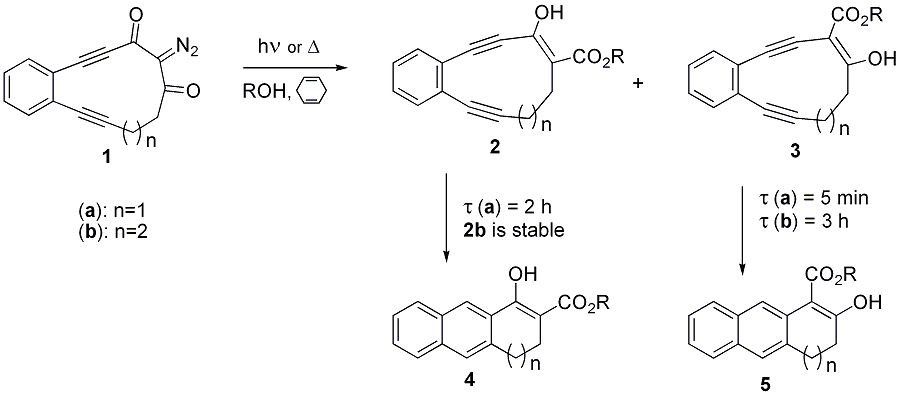
Upon irradiation with 350 nm light 1a,b undergo Wolff Rearrangement resulting in ring contraction and the formation of isomeric enediyne-ketoesters 2 and 3. According to DFT calculations and NMR data for 2b, 3a,b the ketoester moiety is fully enolized. The additional endocyclic double bond enhances the reactivity of these enediynes towards Bergman cyclization, positioning them among most reactive natural, as well as synthetic, enediyne compounds. It is noteworthy that these reactive enediynes are generated from thermally (up to at least 800C) precursors.
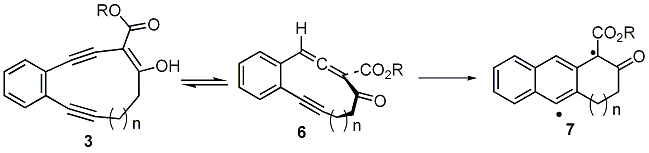
The interesting feature of this system is big difference in reactivity between 2 and 3 (Scheme 1). We hypothesized that this difference stemmed from the ability of 3 to ketonize into allenic isomer 6 (Scheme 2). The latter can undergo more facile Myers cyclization to produce diradical 7.
To test this hypothesis we have prepared an analog of 3a, benzocyclodecaenediyne 8, which contains a carbonyl moiety separated by methylene group from acetylenic terminus (Scheme 3).

Enediyne 8 is a surprisingly reactive substance. Upon gentle heating, in the presence of traces of acid or bases it undergoes rapid cycloaromatization to anthracene derivative 12. The weak specific acid, as well as strong general base, catalysis, and pronounced solvent isotope effect in normal direction indicate that the rate-limiting step of this process is enolization of 8 to 9. The latter rapidly tautomerize to the allenic ketonone 10, which then undergoes facile Myers cyclization.
In summary, we have developed method of the photochemical generation of reactive enediynes from thermally stable precursors. We currently use this discovery in design of advance photoswitchable enediynes.