
ACS PRF | ACS
All e-Annual Reports

43443-AC1
Catalytic and Potentially Enantioselective ERC and EHC Reactions
I. Improved diastereoselection in electroreductive cyclization reactions. We previously defined the term "electroreductive cyclization" (ERC) to refer to the electrochemically initiated reduction and subsequent intramolecular cyclization of an electron deficient alkene onto a remotely tethered aldehyde or ketone. Since the first examples of the electrochemical process were published, researchers have developed many non-electrochemical alternatives to accomplish the same bond construction. Unfortunately, the electrochemical protocol often suffers from low stereoselectivities.
As illustrated in Tables 1 and 2, the diastereoselectivity of electroreductive cyclization reactions can be improved significantly by using lanthanide ions or Mg2+ that is electrochemically generated at a sacrificial anode. The ions presumably serve to coordinate with the Lewis base sites found at each end of the substrate chain, thereby establishing two complexes of differing energies. The diastereoselectivity most likely arises as a consequence of competition between transition states that resemble structures I.4 and I.5. While it was not possible to affect any of the processes catalytically, our search for a suitable mediator continues.
Table 1. The influence of ytterbium ions upon the diastereoselectivity of ERC reactions.
substrate additive yield trans:cis** (% de) I.1 a none 79-86 1 : 1 (0) I.1 a 1 equiv Yb(OTf)3 84 4.5:1 (64) I.1 b none 83-87 1 : 1 (0) I.1 b 1 equiv Yb(OTf)3 81 6:1 (72) 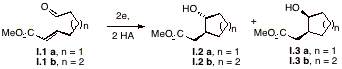
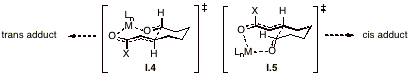
Table 2. The effect of Mg2+ upon the diastereoselectivity of ERC reactions.
Substrate
| additive
| yield
| trans:cis (%de)
|
I.1 a
| none
| 79-86
| 1 : 1 (0)
|
I.1 a
| Mg2+
| 84
| 6 : 1 (72)
|
I.1 b
| none
| 83-87
| 1 : 1 (0)
|
I.1 b
| Mg2+
| 82-89
| 8-9 : 1 (78-80)
|
The catalytic electrochemically mediated oxidation of housane II.1 leads to a cation radical that engages in a rearrangement leading cleanly to the (4.3.0) adduct II.2. The appearance of a catalytic current in the cyclic voltammogram of a solution containing the tris(aryl)amine and housane II.1 indicated that the amminium cation radical II.3 ![]() is able to oxidize the housane and return the mediator to the original redox couple. DFT calculations show electron density on both the aryl and strained sigma framework in the HOMO of housane II.1. From the spin density and electrostatic potential map for the cation radical emerges a picture where the spin resides on the side that is distal to the substituent, while the hole is proximal to it. Both experiment and theory show that the rearrangement is best characterized as a [1,2]-carbon shift toward an electron deficient site and that migration toward the substituent-bearing carbon is much preferred over the alternative pathway.
is able to oxidize the housane and return the mediator to the original redox couple. DFT calculations show electron density on both the aryl and strained sigma framework in the HOMO of housane II.1. From the spin density and electrostatic potential map for the cation radical emerges a picture where the spin resides on the side that is distal to the substituent, while the hole is proximal to it. Both experiment and theory show that the rearrangement is best characterized as a [1,2]-carbon shift toward an electron deficient site and that migration toward the substituent-bearing carbon is much preferred over the alternative pathway.
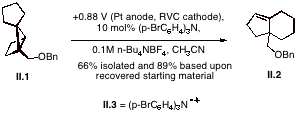
The insight gained during these investigations promises to be of utility in designing substrates that will allow the housane-derived cation radical rearrangement reaction to be applied to the synthesis of specific structures, particularly, to those of natural products. Our efforts to apply the chemistry in this manner are underway. III. Impacts. The chemistry described in part I was carried out by several graduate students. For Jennifer Mallory, it forms the basis for her ongoing studies toward the PhD degree, as she prepares to utilize the results in the synthesis of a natural product. The research described in part II forms a significant portion of the PhD research of Young Sam Park (graduate student). Its broad scope (synthesis, mechanism, calculations, electrochemistry, catalysis) continues to provide an exceptionally rich training ground. The work remains a fruitful collaboration between my group and that of Professor Dean Tantillo at UC Davis.