ACS PRF | ACS
All e-Annual Reports

44734-AC1
Polyphospholes and Hetero Analogs
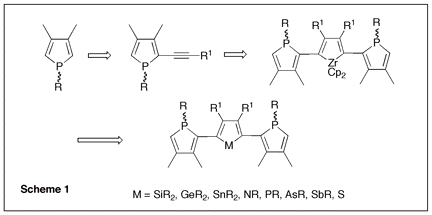
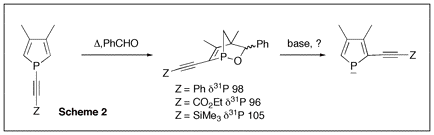
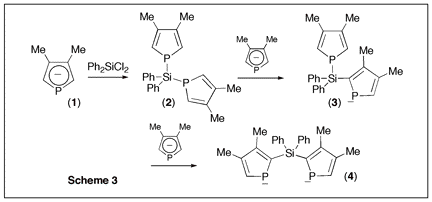
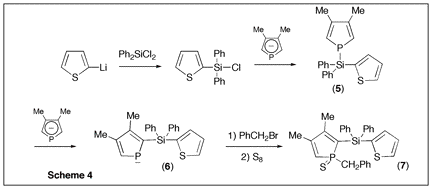
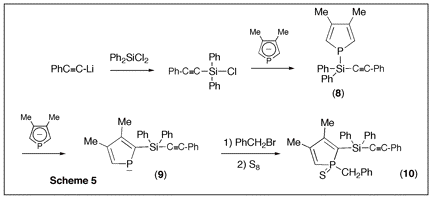
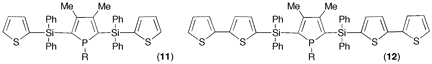
Since phospholes are not aromatic, the main synthetic problem that is encountered when developing their chemistry is the lack of direct functionalization methods. The philosophy of the proposed research program was to use a recently developed phospholide functionalization technique1 to prepare 2-alkynyl substituted phospholes and then use zirconacyclopentadiene chemistry to convert these alkynes into phosphole-heterole-phosphole oligomers of potential use in optoelectronics. (Scheme 1) Initial attempts The only simple access to 2-functional phospholes relies on the [1,5] shift of the functional group from P to C.2 In the case of alkynyl groups, the migration is difficult (barrier ca. 30 kcal mol-1)2 and the CĽC triple bond can react with the intermediate 2H-phosphole to give polymers. After several attempts, we found that it is possible to trap the alkynyl-substituted 2H-phosphole as a [4+2] cycloadduct with benzaldehyde. (Scheme 2) Unfortunately all our attempts to perform the cycloreversion with a base (as described in the non-functional case3) to get the alkynyl-substituted phospholides failed. Introducing silicon Silicon has two interesting properties: first, it easily migrates (the barrier for SiH3 is only 14 kcal mol-1)2; besides, it transmits the conjugating effects through its low-lying empty orbitals.4 Replacing the direct alkyne-phosphole by an alkyne-silicon-phosphole link was thus a possible option. The problem was that the P-Si bond is easily cleaved by oxygen bases. We thus decided to investigate the silicon shift in the presence of other bases than the normally used tBuOK. Since silicon groups are electron-withdrawing, we reasoned that the starting non-functional phospholide is just strong enough as a base to deprotonate the functional 2H-phospholes. It is, of course, fully compatible with the P-Si bond. We decided to check this idea with the synthesis of a Ca-SiR2-Ca bridge between two phosphole rings (Scheme 3). The protonated phospholide (1) was recovered as the 2H-phosphole [4+2] dimer (d31P -- 64.4 and Đ 25.2 ppm, 3JPP = 182 Hz).5 The final dianion (4) was unambiguously characterized by 31P NMR (d31P + 111 ppm) and anionic mass spectrometry. The phosphole-silicon-phosphole link proved to be sensitive to hydrolysis and we did not pursue in this direction. We then envisaged to use silicon as a carrier for a thiophene ring. The [1,5] shift of thiophene around the phosphole ring needs strong heating6 and we thought that the synthesis of a a-connected phosphole-silylene-thiophene chain under mild conditions would be worthwhile. Indeed, both phosphole-thiophene7 and thiophene-silylene8 mixed chains have proven their worth for the manufacture of OLED's or organic transistors. The one-pot reaction was carried out as shown in scheme (4). All of the transformations take place at room temperature. The functional phospholide was characterized by its 31P resonance at + 105.4 ppm. It was fully identified as its P-benzyl sulfide (7) that was characterized by X-ray crystal structure analysis. This approach to silyl-functional phospholes can be generalized. Our next logical step was to use silicon as a carrier for an alkynyl group (Scheme 5). The 31P resonances of (8) and (9) appear at -59.2 and +104.4 ppm, respectively. We tested the feasibility of the zirconium coupling reaction on (10). Zirconocene (from Cp2ZrCl2 and BuLi) reduces the P=S bond but does not give useful adducts with the CĽC triple bond. Before closing this section, we have to mention that this phospholide-based methodology can be used to perform single or double [1,5] shifts of phosphino groups around the phosphole ring leading to original a-connected phosphine-phosphole-phosphine tridentate ligands of interest for catalytic studies. We are preparing a publication describing our results with silicon and phosphorus migrating groups. Defining new targets On the basis of this new methodology, we propose to synthesize the following original chains (11-12). Transposition with N-methylpyrrole appears possible. Since the lithium derivative of TTF is easily accessible,9 the synthesis of TTF-silylene-phosphole-silylene-TTF also appears possible. All of these chains look exciting from the standpoint of applications in optoelectronics. References 1) Holand, S.; Jeanjean, M.; Mathey, F. Angew. Chem. Int. Ed. Engl. 1997, 36, 98. 2) Mathey, F. Acc. Chem. Res. 2004, 37, 954. 3) Toullec, P.; Ricard, L.; Mathey, F. J. Org. Chem. 2003, 68, 2803. 4) Fujitsuka, M.; Cho, D. W.; Ohshita, J.; Kunai, A.; Majima, T. J. Phys. Chem. B 2006, 110, 12446. 5) Charrier, C.; Bonnard, H.; De Lauzon, G.; Mathey, F. J. Am. Chem. Soc. 1983, 105, 6871. 6) Bevierre, M. O.; Mercier, F.; Ricard, L.; Mathey, F. Bull. Soc. Chim. Fr. 1992, 129, 1. 7) Su, H.-C.; Fahdel, O.; Yang, C.-J.; Cho, T.-Y.; Fave, C.; Hissler, M.; Wu, C.-C.; RŽau, R. J. Am. Chem. Soc, 2006, 128, 983. 8) Kim, D.-H. ; Ohshita, J. ; Kosuge, T. ; Kunugi, Y. ; Kunai, A. Chem. Lett. 2006, 35, 266. 9) FourmiguŽ, M.; Huang, Y.-S. Organometallics 1993, 12, 797.