www.acsprf.org
Reports: DNI148864-DNI1: Exploring the B-N/C=C Isosterism in Carbon-Rich Compounds
Exploring the B–N/C=C Isosterism in Carbon-Rich Compounds
Abstract
The C=C bond unit is a ubiquitous and fundamentally important structural motif in organic chemistry. The substitution of the organic C=C unit with the isosteric inorganic B–N bond in organic molecules can lead to novel hybrid structures with unique properties. The proposed research seeks to illuminate the B–N vs. C=C isosterism in conjugated carbon-rich compounds by investigating the effects of this isosterism in benzene derivatives, i.e., 1,2-azaborines. We have accomplished the synthesis of a versatile intermediate for the construction of 1,2-azaborine-based conjugated scaffolds. We also accomplished the synthesis, structural characterization, and optoelectronic properties of BN-Tolan and Bis-BN-Tolan, and BN-bis(phenethynyl)benzene. The accomplished work sets the foundation for further developments of BN heterocycles in materials science.
Specific Aims
This proposed research seeks to study the B–N vs. C=C isosterism in conjugated structures incorporating the 1,2-azaborine core (Scheme 1). Our goals were: 1) to develop a synthesis of a versatile 1,2-azaborine precursor amenable to substitution at boron, nitrogen, and carbon positions, 2) to construct a series of phenylacetylene derivatives with B–N substitution at various positions, and to 3) to characterize the newly prepared compounds.
| Scheme 1.
|
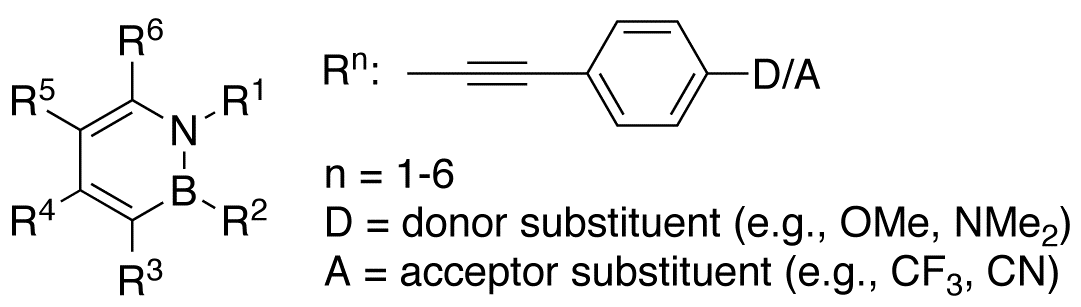 |
Accomplishments
The proposed research is strongly
dependent on the successful synthesis
of the universal precursor 1 bearing three orthogonal functional features
(Scheme 2, top). The strong dependence of the success of the
proposed research on a single crucial target compound has been pointed out by
several reviewers of the proposal. Cognizant of the importance of
compound 1, we pursued its synthesis prior to ACS-PRF funding.
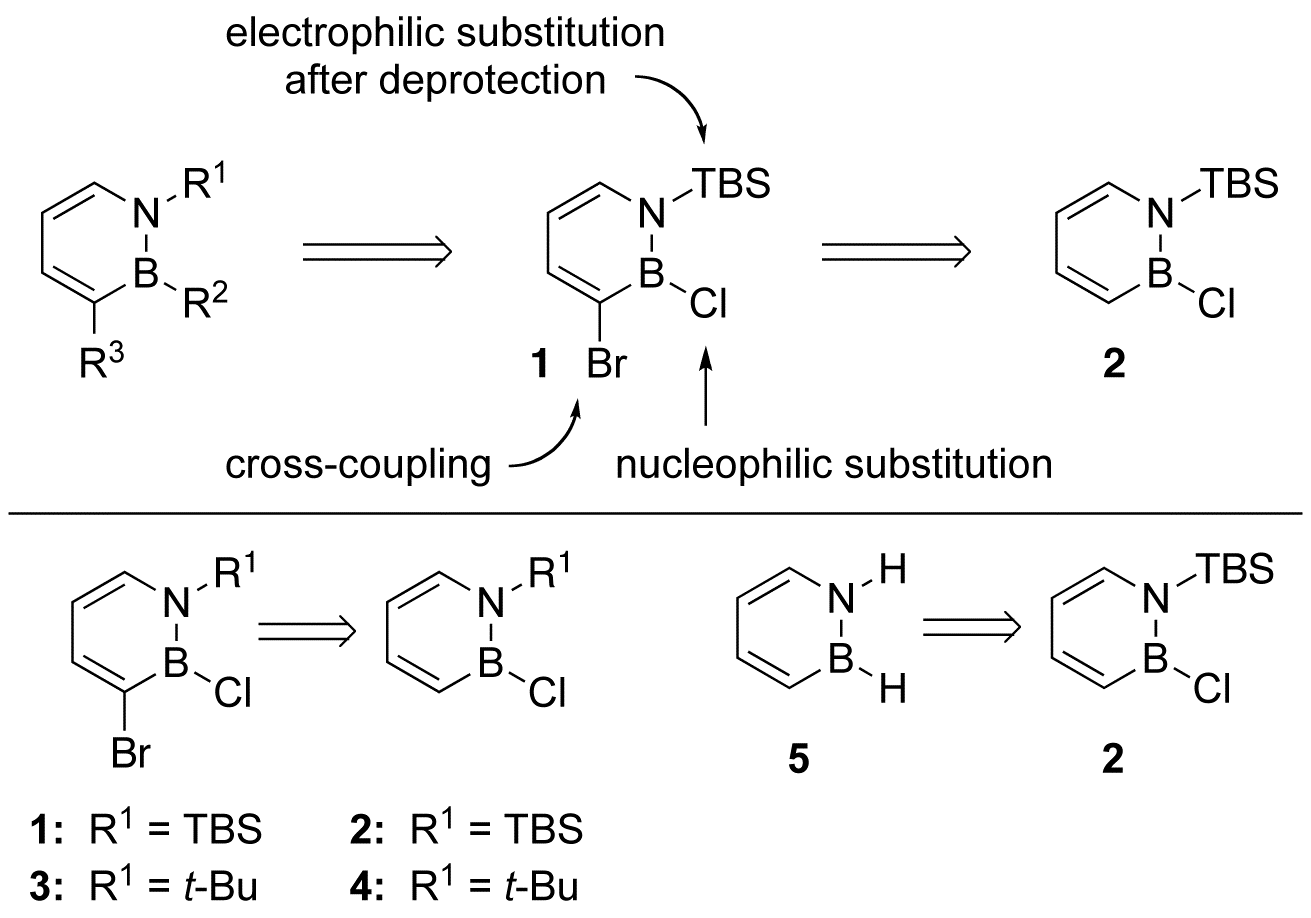
We determined that treatment of 1,2-azaborine 2 with elemental bromine gave
the desired compound 1 in 61% isolated yield (Scheme 2, bottom). Compound 1 has been
characterized by 1H, 13C, 11B NMR, IR, and
high-resolution mass spectroscopy. The related compound 3
has been prepared in the same manner from precursor 4,
the procedure for which has been published in Mol. Biosyst. 2009,
5, 1303-1305. We also determined that the N-TBS can be subsequently removed, which ultimately led to the
first successful isolation and characterization of the parent 1,2-Dihydro-1,2-azaborine 5 (Scheme 2,
bottom, Angew. Chem. Int. Ed. 2009, 48,
973-977). Scheme 2.
Having versatile synthetic precursors 1 and 2 available, and with the help of ACS-PRF funding, we described in the last reporting period the synthsis and characterization of BN-substituted diphenylacetylene (tolan) as our initial conjugated system to investigate because of its structural simplicity and its significance in materials science. Diphenylacetylene serves as an important building block in liquid crystals and is the fundamental sub-unit in the structure of the carbon allotrope graphyne.
We synthesized "BN-doped" tolan derivatives BN-Tolan and Bis-BN-Tolan (Scheme 3). We determined that Bis-BN-Tolan displays unique N–Háááp(C¼C) hydrogen bonding in the solid state consistent with the N–H moiety possessing a hybrid character between an arene C–H and an amine N–H bond. This bond energy is substantial, on the order of 5 to 6 kcal/mol. Furthermore, we established that BN tolans display absorption and emission spectra that are distinct from their carbonaceous analogue. Time dependent-DFT calculations show that for BN-tolans and tolan, the singlet excited state transitions are dominated by the HOMO-LUMO transition with little charge transfer character consistent with the observed solvatochromism studies.
| Scheme 3.
|
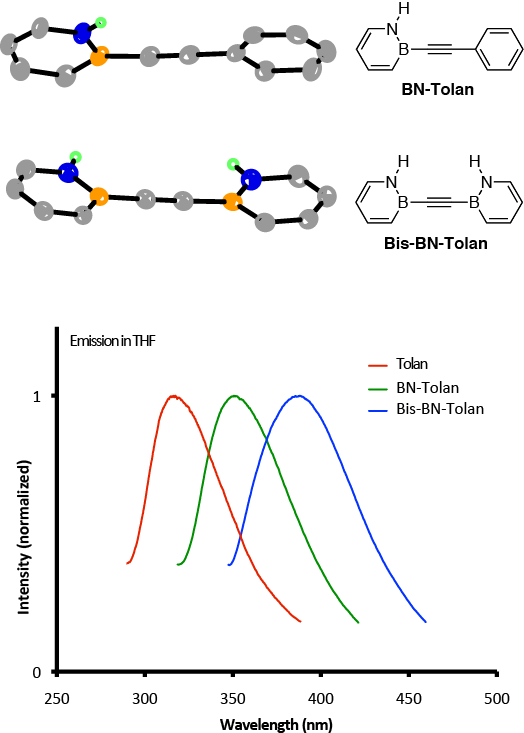 |
| Scheme 4.
|
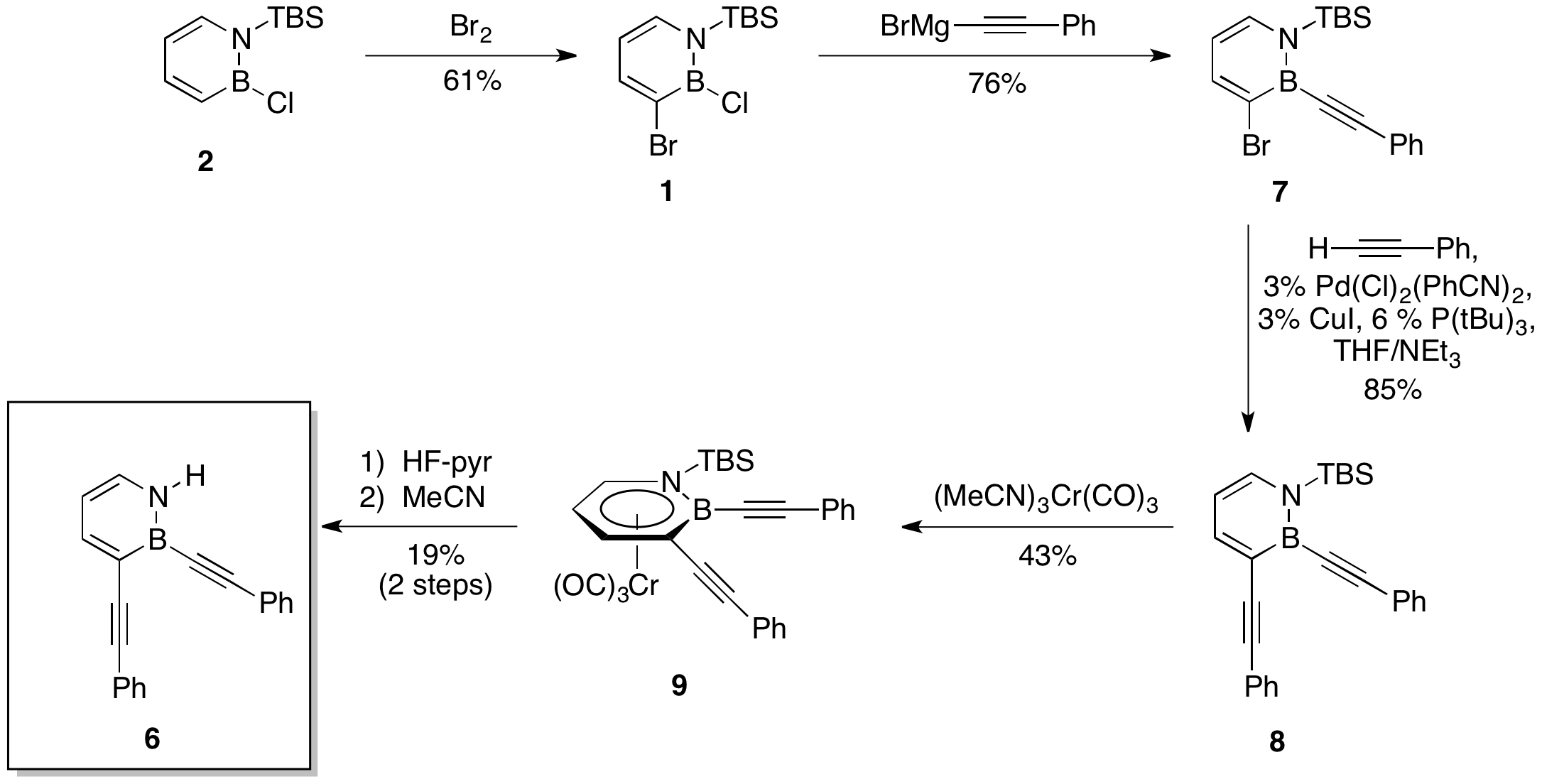 |
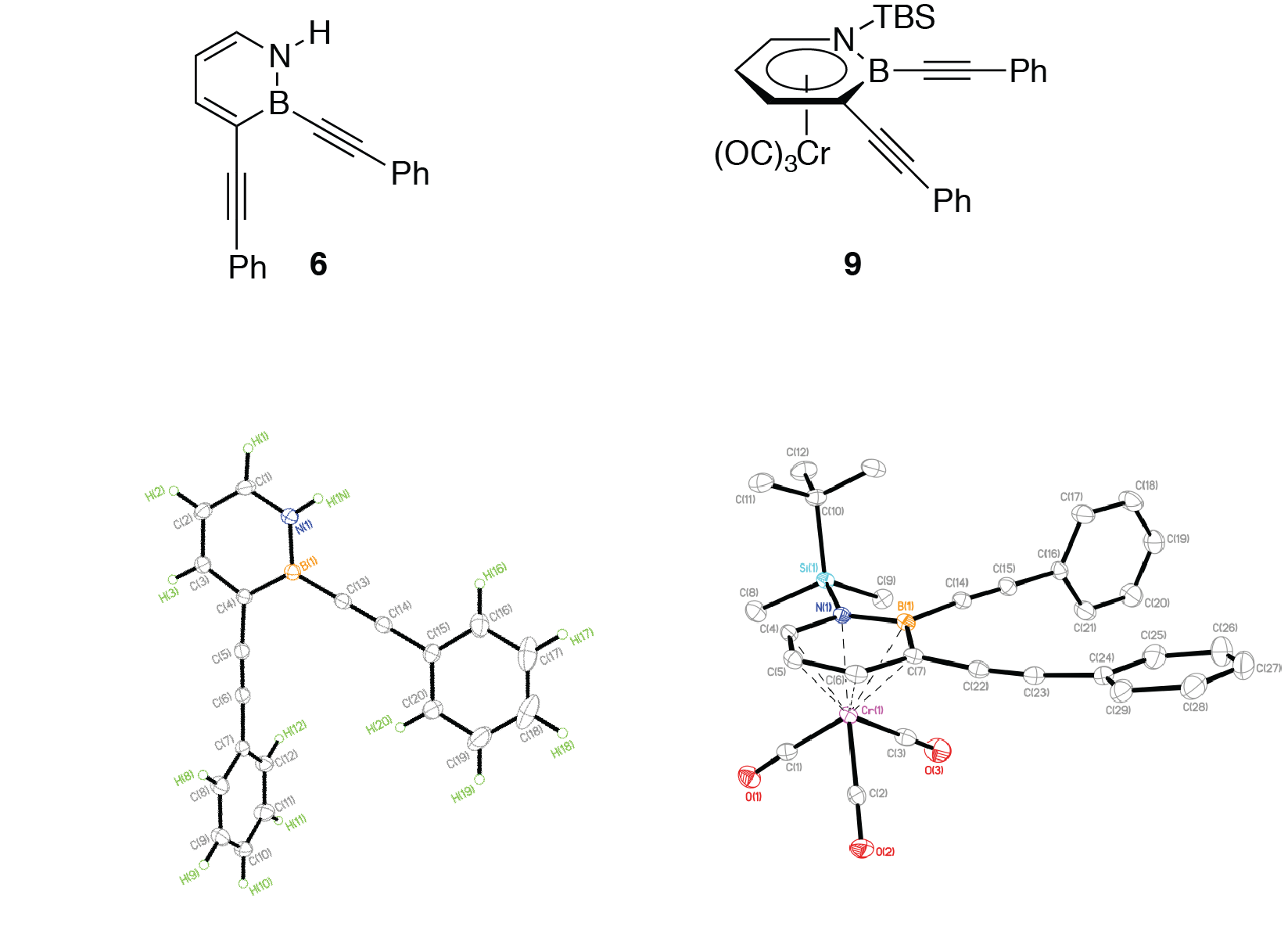
In this reporting period, we have started to target more conjugated systems linked by the 1,2-azaborine core using precursor 1. Specifically, we prepared the parent BN-bis(phenethynyl)benzene 6 and characterized its absorption and emission properties. The synthesis of diyne 6 is shown in Scheme 4. Bromination of B-Cl 1,2-azaborine 2 with Br2 proceeded exclusively at the C3 position, providing 1 in good yield. Nucleophilic substitution of 1 with phenylethynylmagnesium bromide provided alkyne-substituted 7 in 76% yield. The lynchpin of our synthesis was the Sonogashira cross-coupling of 7 with phenylacetylene, which gratifyingly produced diyne 8 in 85% yield. It is noteworthy that this is the first demonstration of a transition metal-catalyzed cross-coupling reaction of a 1,2-azaborine. We were unable to directly cleave the TBS protecting group from diyne 8. Instead, the complexation of 8 with tricarbonylchromium(0) trisacetonitrile provided piano-stool complex 9 in 43% yield. The identity of complex 9 was confirmed by single-crystal X-ray crystallography. The N-TBS group of 9 was cleaved with HF-pyridine. The reaction mixture was directly added to MeCN, whereupon diyne 6 was isolated via silica gel chromatography, albeit in low yield.
We have characterized 6 by NMR, IR, UV-Vis, and fluorescence spectroscopy, as well as high-resolution mass spectrometry (HRMS), all of which are consistent with the assigned structure. The structure of 6 has been confirmed by single-crystal X-ray crystallography. Scheme 5 illustrates the ORTEP diagrams of compounds 6 and 9.
| Scheme 5.
|
 |
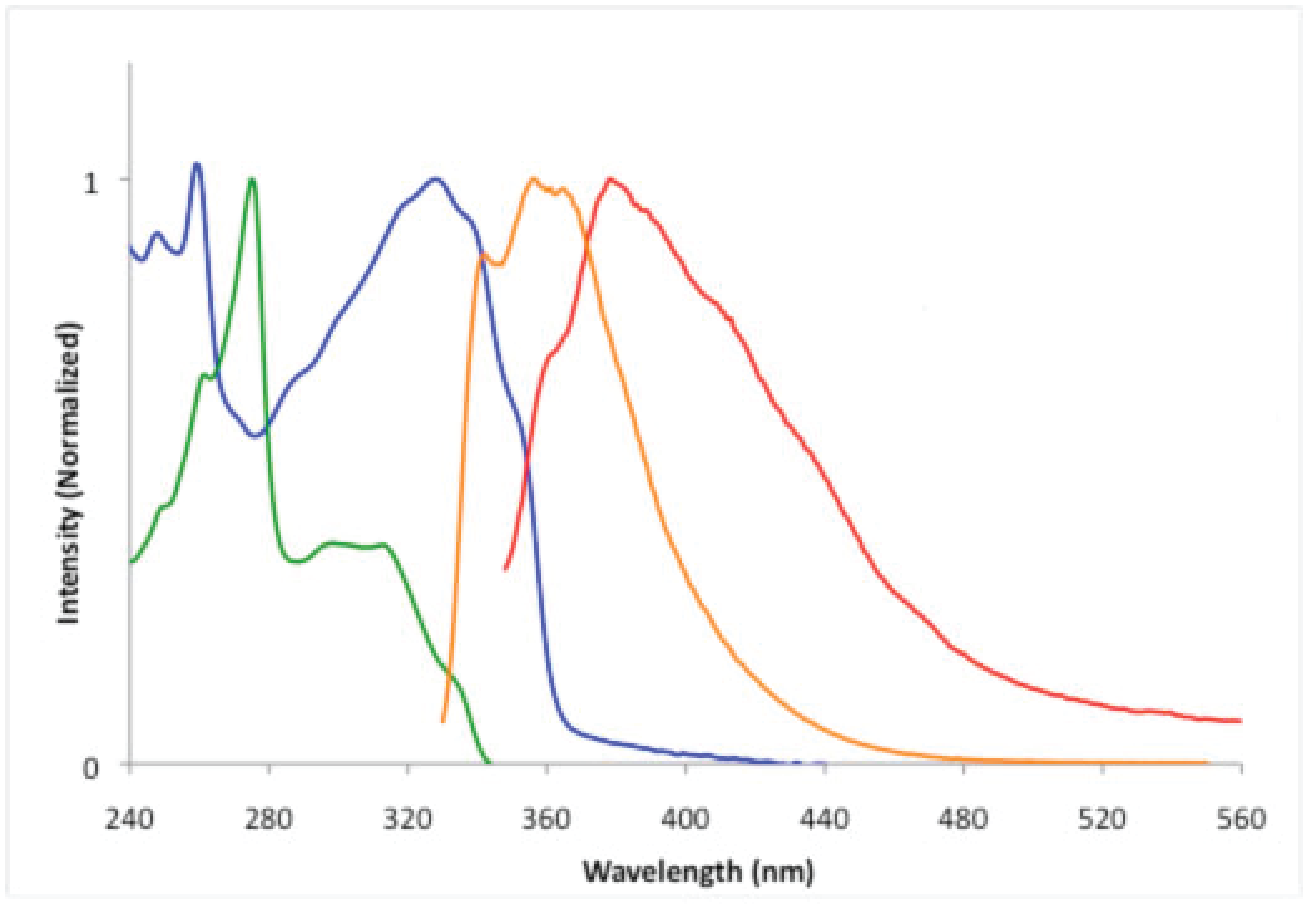
The absorption and emission spectra of 6 are shown in Scheme 6, along with the absorption and emission spectra of 1,2-bis(phenylethynyl)benzene. The absorption spectrum of 6 (blue trace) shows a broad absorption maximum at 328 nm (e = 23316 M-1cm-1). By comparison, the absorption spectrum of 1,2-bis(phenylethynyl)benzene (green trace) shows an absorption at 315 nm (e = 10723 M-1cm-1). Therefore, the HOMO-LUMO transition appears quite similar in 6 and the carbon-based analog. The fluorescence spectrum of heterocycle 6 (red trace) displays a peak at 378 nm (FPL = 0.08). In contrast, 1,2-bis(phenylethynyl)benzene (orange trace) shows an emission at 356 nm (FPL = 0.24). The bathochromic shift observed for compound 6 relative to its carbon analog is consistent with our previous study of BN-doped analogs of diphenylacetylene.
| Scheme 6.
|
Current work are focused on developing donor-acceptor-substituted conjugated systems linked by the BN-heterocyclic core. The ACS-PRF support has provided the foundation for the development of BN-heterocycles in potential optoelectronic materials applications.