www.acsprf.org
Reports: UR150086-UR1: Oxidation of Alkenes in Aqueous Solvent Mixtures Using Environmentally Benign Reagents
Research in our laboratory is aimed at developing eco-friendly protocols for oxidative transformations of alkenes. In particular, we are interested in carrying out oxidative transformations in aqueous solvent mixtures and using water soluble using benign hypervalent iodine reagents.
Last year we noted that the readily formed iodonium ion, 2 , from the oxidation of 4-iodobenzoic acid, 1 , is capable of effecting oxidative cleavage of alkenes. We also ascertained that oxidative cleavage could be carried out successfully using catalytic amounts of 2 as long as stoichiometric amounts of the co-oxidant, Oxone was present in the reaction. An extensive substrate scope investigation was also part of the initial study.
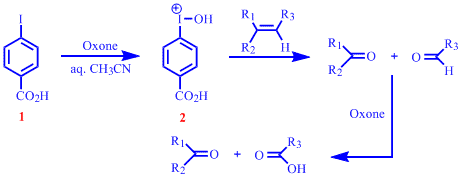
A significant amount of our effort during the current project year was directed at understanding the mechanism of this reaction. A variation in the rate of cleavage of different substrates was noted during the substrate scope study. We wanted to mechanistically justify the observed rate differences. In general, it was found that the cleavage of phenyl conjugated cyclic alkenes were considerably faster than phenyl conjugated acylic alkenes. Non-phenyl conjugated alkenes (both cyclic and acyclic) cleaved the slowest. The ability to use vicinal diols as substrates was also viewed as an opportunity to gain further insight into plausible reaction mechanism. To rationalize some of the trends observed we performed a series of competitive oxidative cleavage experiments. Figure below shows a group of alkenes and vicinal diols used in the head-to-head competitive oxidative cleavage reactions.
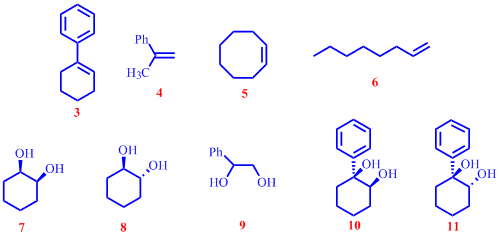
Monitoring the competitive oxidation reactions by 1H NMR showed that 3 was cleaved faster than 4 and the rate of cleavage of both 10 and 11 were faster than that of 7 , evidently indicating the favorable effect of having a phenyl group on the bond being cleaved. We believe that the stability afforded by the phenyl ring on any build up of positive charge is responsible for observed rate differences. More clues about the reaction mechanism came from the studies of the cleavage of vicinal diols. During optimization studies reported last year it was observed that cis-diol 10 was cleaved faster than trans-diol 11 and we were curious whether such an observation offered an insight into a possible reaction mechanism. Head-to-head competition experiments between 7 and 8 clearly demonstrated that cis-1,2-cyclohexanediol, 7 , was cleaved faster than its trans counterpart, 8 . In comparing rates of cleavage of a mixture of 10/11 and 7 it was noted that even trans diol 11 cleaved faster than 7 , underscoring the importance of the phenyl group in presumably stabilizing an incipient carbocation. Additionally, competition experiments between 10/11 and 9 showed that 10 and 11 cleaved faster than 9 , suggesting that conformationally rigid cis diols are better substrates than open chain vicinal diols. No discernible rate differences in the cleavage 7 and 9 were noted. This latter observation suggests that the conformational rigidity of the cyclic cis diols offsets the favorable role of a phenyl group on the bond cleaved in acylic substrates. Based on these studies, the following mechanism for the oxidative cleavage of alkenes is proposed, using the cleavage of 1-phenyl-1cyclohexene, 3 , as an example.
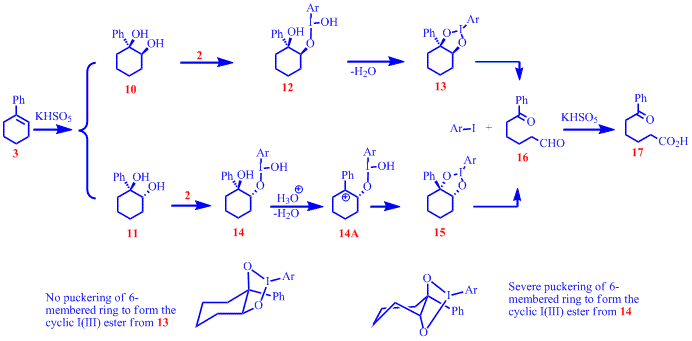
The acidity of the reaction medium (pH< 2.0) due to the presence of Oxone as the co-oxidant precludes its use for the cleavage of substrates that carry acid sensitive functional groups. One of our pressing goals is to identify an alternate and non-acidic co-oxidant for the reaction. We also plan to identify a suitably buffered reaction medium to successfully carry out the oxidative cleavage reactions.
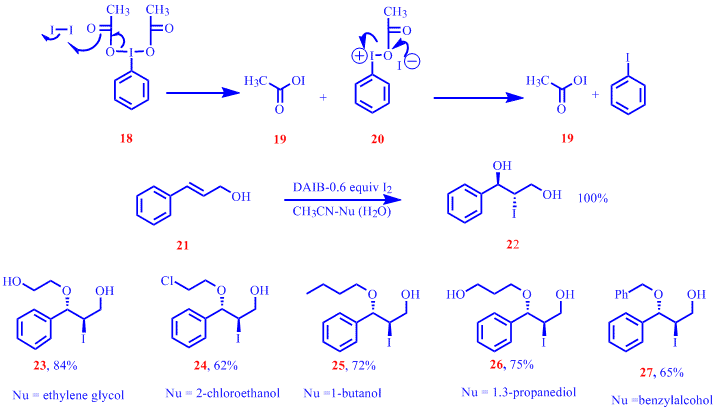
Last year we also reported a versatile and an iodine atom economic protocol for co-iodination of alkenes. Oxidation of I2 using (diacetoxy)iodobenzene (DAIB), 18 and the trapping of the iodide ion by 20 to generate a second equivalent of acetyl hypoiodite as shown in Scheme below was unequivocally demonstrated by 1HNMR from the quantitative conversion of cinnamyl alcohol, 21 to the corresponding iodohydrin 22 . Also shown was the successful use of a variety of nucleophilic additives in the co-iodination of cinnmayl alcohol to rapidly generate multifunctionalized derivatives such as 23 - 27 .
Using the optimized reaction conditions we extended the substrate pool to include a diverse array of alkenes to verify the generality of the method and the results were reported last year. Two curious observations made from our initail susbstrate scope investigation are shown below. While the electron deficient alkene 28 failed to react , the attempted co-iodination of 29 provided only 30 in 72% yield establishing a selectivity of the procedure for electron rich alkenes. The use of [bis(trifluoroacetoxy)iodo]benzene (BTI), 31 , as the hypervalent iodine oxidant in the coiodination reactions of 28 and 29 provided products from the functionalization of electron electron deficient double bonds also. This interesting observation and the potential for product control by varying the oxidant hinted
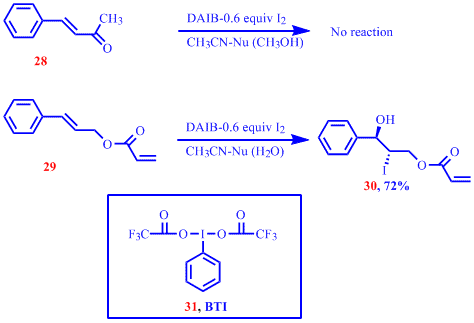
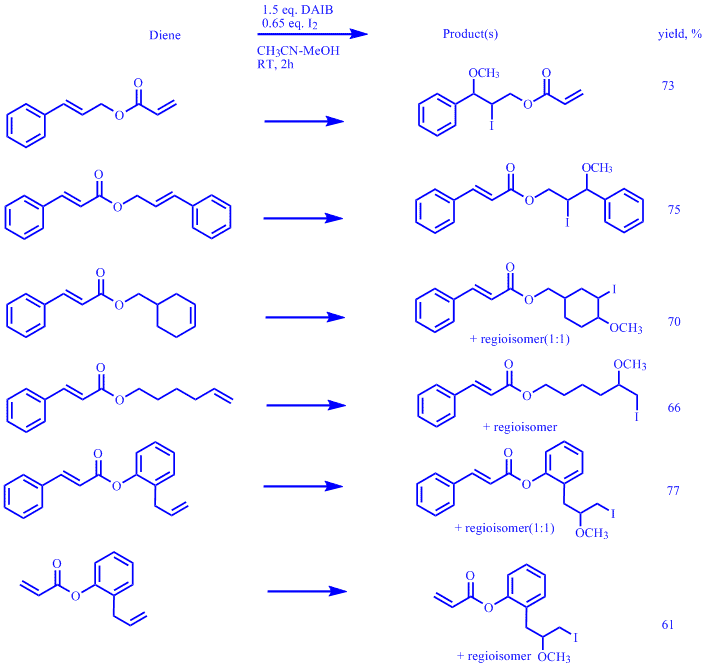
the possibility of differently functionalizing dienes bearing both electron deficient and electron rich double bonds. Prior to embarking on this interesting mini project we further investigated the selective co-iodination of electron rich double bonds in dienes using DAIB as the oxidant and the results are shown in the Table below.
Additionally, we have also demonstrated that dienes can be differently functionalized by careful choice of the oxidant and the nucleophilic additives in the reaction. Synthesis of isomeric derivatives 34 and 36 can be readily achieved from 32 with proper choice of nucleophilic aditives in sequentially carried out co-iodination reactions.
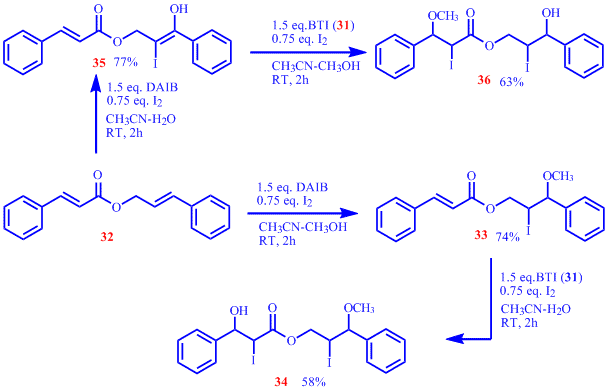
In conclusion, we have developed a versatile and iodine atom economic co-iodination protocol for alkenes. The two C=C bonds of dienes can be selectively functionalized by careful choice of the oxidant and the nucleophilic additive in the reaction as demonstrated in the conversion of 32 to 34 and 36 respectively. We are currently exploring the selective functionalization of the double bonds in dienes using the developed co-iodination protocols.