www.acsprf.org
Reports: UNI150337-UNI1: Development of a Facile Tandem Asymmetric Epoxidation/Ring-Opening Protocol: An Efficient Entry to Enantiomerically Enriched Tertiary Naphythoquinols
Our proposal had two primary goals: (1) to develop a highly efficient method for the construction of either enantiomer of chiral naphthoquinols via a tandem oxidation/ring opening of cyclic 3,4-epoxyalcohols; (2) to employ the proposed methodology in conjunction with a tandem intramolecular biaryl ether formation/phenolic oxidation sequence to access the core framework of the natural products spiroxins A-E in an asymmetric fashion. During the past year, we have successfully completed the first goal of the project, finalized all the data collection for this goal and prepared a manuscript that has been published in Tetrahedron Letters.
In 1999, the natural product spiroxin A was isolated from a marine fungus (strain LL-37H248) and it was found to exhibit antitumor activity against human ovarian carcinoma in mouse xenografts at a 1mg/kg dose.1 The important biological properties of spiroxin A, and its fascinating molecular structure, prompted my research lab to design a novel synthetic approach towards this promising anticancer natural product. Our approach towards the total synthesis of the natural product proceeds through a key chiral tertiary naphthoquinol intermediate followed by the construction of the octacyclic core. The key tertiary naphthoquinol, besides being an important synthetic intermediate for the synthesis of spiroxin A, is also present in numerous other natural products with important biological activities. In spite of this fact, almost no methods exist for the asymmetric preparation of quinols from achiral precursors. In this past year, we have focused on developing a highly efficient and facile method to prepare chiral naphthoquinols.
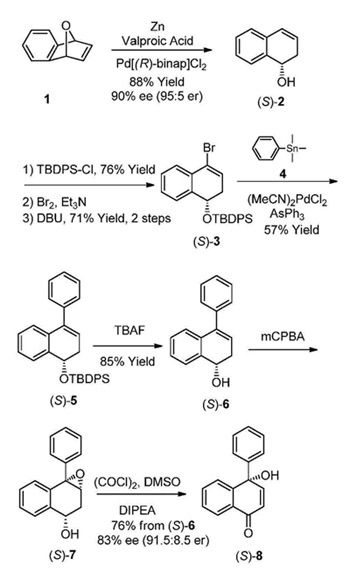
The proposed methodology project (Goal 1) consisted of a series of seven synthetic steps. During this past year, we have completed all of the proposed synthetic steps, have unambiguously characterized all of the synthetic intermediates and optimized several intermediate steps. Validation of the tandem oxidation/ring-opening methodology was performed using the (S)-enantiomer of an homoallylic alcohol as a model substrate. For Goal 1, reaction conditions using the homoallylic alcohol (S)-2 were developed resulting in the formation of the desired tertiary naphthoquinol product (S)-8 with high efficiency and enantioselectivity. The homoallylic alcohol (S)-2 was prepared by a stereoselective ring opening of cyclic ether 1 using Pd[(R)-binap]Cl2, zinc and valproic acid in 88% yield. This critical step allows for the enantioselective synthesis of both (+) and (-) tertiary naphthoquinols 8, as either enantiomer is preparable from the single precursor simply by switching the ligand. The protection of homoallylic alcohol (S)-2 was accomplished using tert-butyl diphenylsilyl chloride and imidazole to generate the TBDPS-protected (S)-2 silyl ether in 76% yield. The vinyl bromide derivative (S)-3 was obtained by a bromination/elimination sequence in isolated yields of 76% and 71% respectively. Coupling of the bromide (S)-3 was accomplished using palladium catalyzed Stille cross coupling conditions with trimethyl(phenyl)stannane (4), to successfully afford the desired (S)-5 coupled product in 57% isolated yield. During this year, we also investigated the efficiency of this reaction using Suzuki coupling conditions with phenylboronic acid and Pd(PPh3)4, however the reaction did not always proceed consistently. Intermediate (S)-5 was then deprotected using tetrabutylammonium flouride to give the alcohol (S)-6 in 85% isolated yield. The preparation of the epoxyalcohol (S)-7 by a directed m-CPBA epoxidation of (S)-6 was successfully confirmed by NMR analysis, but all attempts to isolate (S)-7 by flash column chromatography resulted in decomposition. Therefore, a directed m-CPBA epoxidation of (S)-6 followed by an oxidation/ring-opening of the resulting intermediate epoxyalcohol (S)-7 under Swern conditions without purification of intermediates was investigated. To our delight, this reaction sequence readily afforded the desired chiral tertiary naphthoquinol (S)-8 in 76% isolated yield from (S)-6 and 83% ee (91.5:8.5 er). These combined efforts resulted in the development of the first general catalytic asymmetric method to access chiral tertiary naphthoquinols and substituted benzoquinols as highly useful structural building blocks in organic synthesis. The high prevalence of these structurally important moieties renders this method particularly attractive for application in the biologically and medicinally interesting molecules.
So far, we have fully addressed Goal 1 of our proposal and have developed a convenient new strategy for the enantioselective synthesis of tertiary naphthoquinols, through a process based on a facile epoxidation and ring opening sequence. These results were the basis of presentations at the 2011 Wellesley College Ruhlman Conference and the American Chemical Society 241st National Meeting, and a manuscript accepted in Tetrahedron Letters. We will focus our attention on Goal 2 of our proposal for the upcoming year, investigating the application of this new methodology towards the total synthesis of the core framework of the spiroxins. A long term goal of the project is to accomplish the enantioselective total synthesis of the natural product Spiroxin A and to conduct structure activity relationship (SAR) studies in an effort to discover spiroxin A analogues that may possess superior biological activities.
References:
1. McDonald, L. A.; Abbanat, D. R.; Barbieri, L. R.; Bernan, V. S.; Discafani, C. M.; Greenstein, M.; Janota, K.; Korshalla, J. D.; Lassota, P.; Tischler, M.; Carter, G.T. Spiroxins, DNA cleaving antitumor antibiotics from a marine-derived fungus. Tetrahedron Lett. 40, 2489-2492 (1999).
Student Participation:
Johanna Stein, '10: Johanna synthesized and isolated several key intermediates, and optimized several synthetic steps. After working for a few years as a research assistant at Harvard Medical School Affiliate McLean Hospital, Johanna plans to attend Medical School.
Alice Kwan, '11: Alice synthesized, purified and characterized all key intermediates, and optimized all synthetic steps. Alice also played an active role in preparing the manuscript for Tetrahedron Letters. Alice is now at Stanford University pursuing a masters program in education.
Maria Jun, '11: Helped proofreading the galley proofs for Tetrahedron Letters manuscript and has begun studies towards Goal 2 of the proposal.