Reports: G3
46587-G3 Transition-Metal-Mediated Activation of Sulfur Toward CS Bond Formation
1. Introduction. The ACS-PRF grant has enabled us to develop the coordination chemistry of monoanionic N,N-dialkyl-N',N''-diarylguanidinato ligands (L) and to investigate the reactivity of the Iralkene platform [Ir(L)(alkene)2]. While this work was originally aimed at the activation of elemental sulfur, we also have discovered new chemistry in the reactions of these complexes with dioxygen. In particular, our studies led to a new class of stable complexes of high-valent Ir. Two papers detailing this ACS-PRF supported work have been published, thus far.
2. Iridium(I) Complexes Supported by Guanidinato Ligands. Guanidinates are attractive nitrogen-donor ligands, because they can be readily synthesized and coordinate to a wide range of transition metals. Their use as supporting ligands for low-valent transition metal centers, however, was very limited. We have synthesized and fully characterized IrI complexes of a series of bidentate N,N-dialkyl-N',N''-diarylguanidinate anions, {ArNC(NR2)NAr}, to explore the coordination ability of this ligand platform to transition metals in low oxidation states and to probe its utility in tuning the reactivity of the coordinated metal center. Detailed inspection of the structural and 13C NMR data of these complexes revealed that the dialkyldiarylguanidinates can function as strongly donating ligands and may be considered stronger donors than other monoanionic, nitrogen-based scaffolds such as β‑diketiminates and certain tris(pyrazolyl)borates.
The new [Ir{ArNC(NR2)NAr}(alkene)2] complexes undergo facile reaction with O2 under ambient conditions. A systematic investigation of substituent effects revealed that the reaction rates can be altered by the electronic and steric properties of the ligands. With this work, we have expanded the scope of the guanidinato(1) platform in transition metal chemistry and demonstrated the potential that this ligand framework has to offer for facile modulation of the reactivity of transition metal complexes. (Inorg. Chem. 2008, 47, 11461.)
The [Ir{ArNC(NR2)NAr}(alkene)2] complexes also react readily with S8 to form iridiumsulfido species. The optimization of reaction conditions and the characterization of these species are ongoing. Preliminary reactivity studies suggest that the sulfur atoms of these complexes can be transferred to substrates such as phosphines and isonitriles, whereas reactions with halogenocarbons yield organosulfido complexes.
3. Stabilization of High-valent Iridium in a Nitrogen-donor Environment. In an attempt to explore the range of Ir oxidation states that can be supported by our N,N-dialkyl-N',N''-diarylguanidinato ligands, we have developed the synthesis of tris(guanidinato) complexes of IrIII from IrI precursors (Scheme 1). The resulting [Ir{ArNC(NR2)NAr}3] complexes can be reversibly oxidized at unusually low potentials and are air-sensitive. By comparison, the IrIII/IrIV redox potentials of our new complexes are significantly less positive than those of many other IrIII complexes. Even complexes with very electron-rich ligand sets such as cyclometalated tris(2-pyridylphenyl), corrolato and tris(dithiocarbamato) complexes, which all provide a trianionic environment around the Ir center akin to the tris(guanidinato) complexes, exhibit higher potentials.
Scheme 1. Synthesis of IrIII and IrIV Compounds
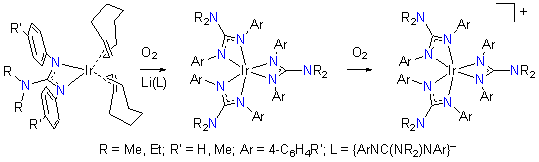
Chemical oxidation of these complexes with O2 or one-electron oxidants such as [FeCp2]PF6 or AgCF3SO3 produced the corresponding IrIV complexes, [Ir{ArNC(NR2)NAr}3]+ (Scheme 1). The oxidized complexes were fully characterized by elemental analysis, ESI mass spectrometry, electronic absorption and EPR spectroscopy, and X-ray crystallography. Based on the experimental data, the complexes are best described as genuine IrIV rather than ligand-oxidized IrIII species. The isolation of stable tris(guanidinato)iridium(IV) complexes is an unusual feature, because these compounds represent the first well-characterized examples of high-valent Ir coordinated exclusively by nitrogen-donor ligands. (J. Am. Chem. Soc. 2009, 131, 9162.)
These results indicate that the dialkyldiarylguanidinato(1) ligands are exceptionally well-suited for the stabilization of transition metals in high oxidation states. Furthermore, the synthesis, interconversion and isolation of complexes in three different oxidation states spanning from IrI to IrIV reveals a remarkable donor flexibility of the guanidinato ligands. Taken together, these properties suggest that the dialkyldiarylguanidinates could be used as ancillary ligands for new complexes that effect multi-electron redox transformations.



