Reports: AC1
48079-AC1 Asymmetric Halogen-Metal Exchange of Geminal Dihalides with Planar Chiral Organometallic Reagents
1. Synopsis of research progress and impact
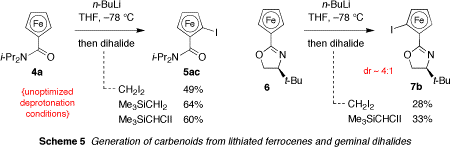
The goal of the funded research is to realize an asymmetric halogen-metal exchange (AHME) reaction of geminal dihalides using a recyclable planar chiral organometallic reagent derived from an iodoferrocene. As originally conceived, the favored reagent design was a 5-substituted 2-iodoferrocene carboxamide (1, YnotH, Scheme 1); however, initial experiments have revealed that such compounds cannot be directly prepared. Alternative AHME reagent possibilities are now being considered; for example, simple 2-iodoferrocence carboxamides 1 (Y=H) and non-carboxamide based systems possessing chiral directed ortho-metallation (DoM) activators. Optimal methods for the preparation of 1,1-diiodoalkanes and 1-chloro-1-iodoalkane AHME substrates have been realized and halogen-metal exchange processes from these geminal dihalides investigated. Studies to date have focused on I/Li and I/Mg exchange of (diiodomethyl)trimethylsilane and (chloroiodomethyl)trimethylsilane. Significantly, it has been established that the carbenoid species derived in this manner add to aldehydes in a Darzens fashion to provide epoxysilane products. This advance constitutes a halogen-metal exchange variant of the Magnus reaction and provides an ideal platform for the immediate future study of AHME. As the second year of sponsored research begins, we are well poised to establish a definitive proof-of-concept for iodoferrocene based AHME.
The positive impact that support from the ACS Petroleum Research Fund has had on my career cannot be overstated. The welcome decision to finance our pilot study of AHME came at a critical juncture when my start-up funds from OSU were all but depleted. The AC grant allowed my research group to continue operating and enabled a talented graduate student, Christopher Emerson, to be relieved of all teaching responsibilities for the first time during his PhD studies. Chris now focuses full time on conducting laboratory research and he is making excellent progress. Results coming within the last few weeks are extremely positive and we believe that will be able to publish preliminary results within the coming year. We also plan on Chris presenting an oral paper at the spring 2010 national ACS meeting. Finally, I am pleased to relay that I was recently successful in securing a four year grant from the NSF to pursue a related research topic concerning carbenoid chemistry. Without PRF support I would not have been able to even reach this point in my career. My prospects for a positive tenure decision next year are now looking very favorable and I am truly grateful to the donors of the ACS PRF for their support.
2. Technical details
2.1. Iodoferrocene synthesis
Ferrocene was converted to tertiary amides 4 (Z=i-Pr, Et) by modified standard methods and subsequent DoM/electrophilic quench provided racemic planar chiral ferrocenes 5 (Y=SiMe3, Me, I) (Scheme 1; nb. Z=Me fails). Suzuki coupling of 5 (Y=I) with phenylboronic acid gave the corresponding phenyl derivatives 5 (Y=Ph). In attempting to transform compounds 5 (Y=SiMe3, Me, or Ph) into the iodoferrocenes of original interest (1, YnotH with X=H), it was discovered that further DoM to the remaining free ortho position is not possible within this series of compounds. Ironically, among the good reasons for setting YnotH (see proposal Section 6.3.2) was a desire to destabilize metallates by exploiting a butressing effect between Y and Z; we speculate that this mechanism is preventing deprotonation and thwarting a direct synthesis of 1 (YnotH). The fully elaborated ferrocene carboxamides 1 (YnotH) are therefore no longer considered as viable reagents for AHME. Fortunately, very recent results suggest that significantly less complex compounds such as 5ac and 5bc are likely to be suitable reagents for AHME (vide infra).
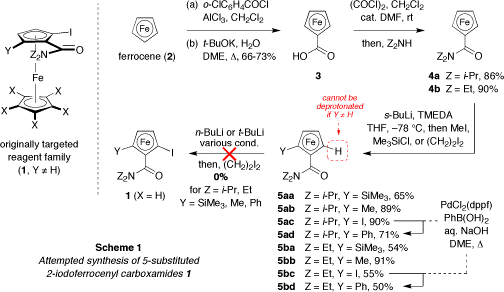
The synthesis of ferrocenes with chiral DoM activators capable of allowing the introduction of a motif similar to that found in our original AHME reagent design 1, is also being explored. Overman and co-workers had already reported one such system for another purpose entirely, the (S)-tert-leucinol derived iodoferrocenyl oxazoline 8, and the preparation of this compound was refined in our laboratory (Scheme 2). While use of reagent 8 will not allow us to probe the effect of pure planar chirality in an AHME process, it does have the advantage that non-racemic material is trivial to obtain. We have also begun to investigate scalemic ferrocenyl sulfoxides (e.g., 11) which may be prepared from Kagan's sulfinate ester 9. An advantage of this compound series is the possibility of obliterating the stereogenic center at sulfur (either by oxidation to a sulfone, or by sulfoxide ligand exchange) to isolate the effect of pure planar chirality on AHME (an inherent feature of the original design 1).
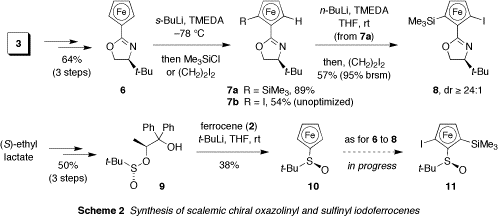
2.2. Halogen-metal exchange studies
Alkyl and silyl substituted diiodomethanes and chloroiodomethanes were prepared using our own modification of the Julia-Charette method (Scheme 3; nb. silyldihalomethanes 13ab were not previously available in pure form). Iodine-metal exchange from these compounds with simple alkylmetals led to carbenoid derived products. After considerable experimentation, it was established that carbenoids generated from 13ab could be reproducibly trapped by benzaldehyde to afford halohydrin or epoxysilane products, but only if oxygen was scrupulously removed from the reaction. Significantly, it was discovered that this halogen-metal initiated variant of the Magnus reaction is completely diastereoselective from the Mg-based carbenoid.
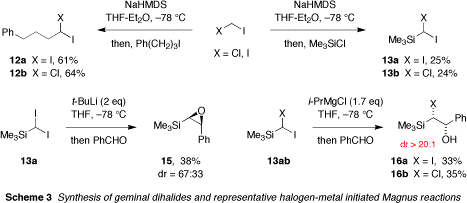
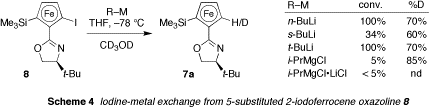
Repeated attempts to initiate the same process from non-racemic chiral iodoferrocene 8, and so realize an enantioselective Magnus reaction via AHME, met with failure. Iodine-lithium exchange from 8 was easily effected (Scheme 4), but the ferrocenyllithium generated in this manner (or via deprotonation of 7a) did not reassimilate iodine when treated with dihalides 12ab or 13ab. The reason for this behavior is not yet understood; however, it was found that protioferrocene 7a was obtained from such reactions when a terminating deuterium quench was performed; possibly indicating proton, rather than iodine, transfer from the dihalomethane.
Attention was recently diverted to DoM activator equipped ferrocenes that lack a butressing element. In both carboxamide and oxazolinyl series, these less elaborate 2-lithioferrocenes underwent successful iodine-lithium exchange with diiodoalkanes and chloroiodoalkanes (Scheme 5). We are in the process of interfacing this chemistry (with metallated ferrocenes generated instead from iodoferrocenes by halogen-metal exchange) with carbenoid trapping experiments to finally establish a proof-of-concept for the proposed AHME cycle.