Reports: AC1
47919-AC1 Development of Catalytic Asymmetric Methods for the Synthesis of Pyrrolidine Derivatives via Hydrogenation of Substituted Pyrroles
Part 1: Stereoselective synthesis of substituted pyrrolidines
Part of the funding provided for this grant period has focused on the heterogeneous catalytic hydrogenation of highly substituted pyrrole systems. These aromatic systems can be fully reduced with excellent diastereoselectivity to afford functionalized pyrrolidines with up to four new stereocenters (equation 1). It is likely that the reaction is a two-step hydrogenation sequence, and that initial reduction of the C=X bond provides a stereocenter that directs the subsequent reduction of the pyrrole.
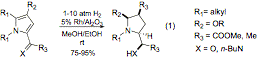
Studies during the reporting period have focused on expanding the scope of the reaction to increase its synthetic value: in order to develop it into 1) a method with broad substrate scope and 2) a practical chemical process. With respect to the first goal, a study focused on expanding the scope of the reaction to nitrogen-containing compounds, which represent an important class of biologically relevant targets. Substrates of types 4, 5 and 6 were chosen for synthesis and hydrogenation studies. Although these studies are not yet complete, we are beginning the process of understanding the relationship between structure and hydrogenation efficiency. For example, with substrate 4 (R=H), we found that hydrogenation was smooth and that the N-N bond was not cleaved under the reaction conditions.
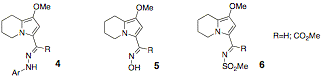
In pursuance of the second goal, we have written an Organic Synthesis article describing the method for use in practical contexts. The chemistry was tested on large scale and documented carefully for this article, so that others may apply the hydrogenation with confidence for their own purposes. Table 1 describes the current scope of the process, and summarizes typical reaction conditions for its practical use in synthesis of racemic pyrrolidine compounds. Using this heterogeneous hydrogenation method, highly substituted pyrroles can be fully reduced with excellent diastereoselectivity to afford functionalized pyrrolidines with up to four new stereocenters. The method is effective for a series of pyrrole a-ketoesters, as shown in Table 1. Typically, the hydrogenation requires 10 atm of H2, 5% (w/w with substrate) rhodium and 12-24 hours of reaction time to ensure complete conversion to the pyrrolidine products 3. Furthermore, it was found that when the hydrogenation in entry 1 was conducted using 1% (w/w with substrate) of Rh/Al2O3 on 0.8 mmol scale, reaction time, selectivity and yield were comparable. If this finding is general, which will be investigated in the next stages of the research, it could represent a significant improvement in cost-effectiveness for the process.
Table 1. Stereoselective Reduction of Bicyclic Pyrrolesa
|
entry
|
pyrrole
|
ratio
|
% yield
|
productc
|
|
1b
|
|
>20:1
|
95
|
|
|
2
|
|
>20:1
|
90
|
|
|
3
|
|
>20:1
|
75
|
|
|
4
|
|
>20:1
|
91
|
|
|
5
|
|
9:1
|
91
|
|
|
6
|
|
>20:1
|
91
|
|
|
7
|
|
>20:1
|
90
|
|
(a) Reaction conditions: 0.1 mmol substrate, 10 atm H2, 5% Rh (w/w with substrate), room temperature, MeOH or EtOH, 12-24 h; (b) Reaction conditions: 0.8 mmol substrate, 1 atm H2, 1% Rh (w/w with substrate), room temperature, EtOH, 6h; (c) Stereochemical assignments are proposed based on the structure of 3a.
In summary, the scope and limitations of the reactions with respect to practical applications were defined more clearly this grant period: for example, identification of optimal catalyst loading, reaction pressure, and expansion of substrate scope was studied. The testing of asymmetric hydrogenation catalysts has also been initiated, although success in this area has been elusive in the short time spent exploring it.
Part 2: The Development of Tandem Annulation Processes via Allenolate Derivatives
Funds during this granting period have also been directed toward another project in the laboratory, focused on a new annulation method. As shown in Scheme 1, β-iodoallenolate intermediates, such as intermediate 8, can be used in annulation reactions to afford products such as cyclohexenyl alcohol 9 and oxadecalin 10.
Scheme 1: β-Iodoallenolate Cyclization
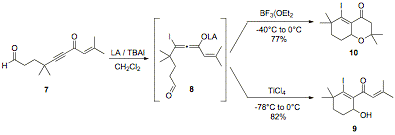
Optimal mono and bicyclization conditions for alkynone 7 were determined by varying the iodide source and the Lewis acid. The results from these reactions indicated the optimal Lewis acid and iodide source for obtaining cyclohexenyl alcohol 9 were TiCl4 and n-Bu4NI (TBAI), and the optimal Lewis acid and iodide source for obtaining oxadecalin 10 were BF3(OEt2 and TBAI, (Scheme 1). The optimal temperature for mono and bicyclization was -78°C to 0°C and -40°C to 0°C, respectively. Furthermore, a stoichiometric amount of BF3(OEt2 was required for the complete conversion of alkynone 7 to oxadecalin 10.
Next, we explored the scope and limitations of both mono and bicyclization. The products obtained from both mono and bicyclization are shown in Chart 2.
Chart 2: Mono and Bicyclization Products
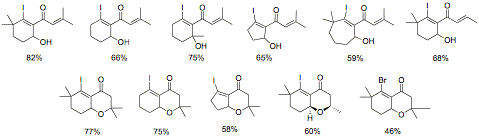
A variety of cyclohexenyl alcohols and oxadecalins were synthesized in moderate to good yields.
The proposed pathway for the conversion of alkynone 7 to oxadecalin 10 is illustrated in Scheme 2. Conjugate addition of the iodide to Lewis acid activated alkynone 7 produces β-iodoallenolate 8. β-Iodoallenolate 8 would then undergo an aldol reaction to produce intermediate 11, which would cyclize to oxadecalin 10 through a Lewis acid triggered intramolecular oxy-Michael addition.
Scheme 2: Proposed Mechanistic Pathway
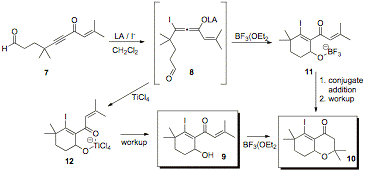
The difference in the reactivity and selectivity that was observed between TiCl4 and BF3(Et2O was likely due to the fact that TiCl4, a bidentate Lewis acid, could be chelated to both oxygen atoms in intermediate 12, which would prevent bicyclization. However, bicyclization can occur with BF3(Et2O, a monodentate Lewis acid, because intermediate 11 is in the proper orientation to allow the oxy-Michael addition and generate oxadecalin 10.
In summary, we have demonstrated the β-iodoallenolate intermediates can be used to produce a variety of cyclohexenyl alcohols and oxadecalins. The success of this β-iodoallenolate cyclization has prompted us to explore its utility in the total synthesis of two biologically active natural products, phomactin A and phomactin D, which is currently underway.



