Reports: G3
46669-G3 Tripodal Phosphido Ligands to Support Early Transition-Metal Catalysis
Part 1. Products of PRF support
PRF support through award 466669-G has resulted in five publications in international journals with additional results that will be reported in at least two additional publications, one to be submitted in the next month and a second likely submitted by the end of the calendar year. The overall result is that PRF support has allowed my research group to grow and be very productive.
Part 2. Impact of PRF support
Over the grant period, the results obtained through PRF support have been vial to the expansion and success of my research group. I have obtained funding from the National Science Foundation, Vermont Space Grant Consortium, Alfred P. Sloan Foundation, and Research Corporation as a result of the momentum that PRF support has helped to establish.
PRF funds have been used to support four different graduate students, two of whom received summer support for two years. These funds released these students from teaching responsibilities and allowed them to pursue work relevant to the grant at 100% effort. This experience greatly improved their development as scientists. Two undergraduate students were supported on the grant, one of whom is a co-author on two publications and is currently pursuing graduate study in chemistry.
The grant has been an important part of establishing my laboratories with respect to results, student training, and extramural funding.
Part 2. Research progress
As noted in the report last year, our initial efforts into tripodal phosphorus ligand preparation has been stymied by synthetic difficulties. In our hands, ligand preparations gave limited reproducible results. The difficult synthesis suggests that these ligands are not currently suited for catalytic studies. In an effort to make more significant research progress, I shifted our efforts toward catalytic chemistry, the heart of the proposed research, using more readily prepared triamidoamine ligands. We had great success looking into bond-forming catalysis involving arsenic, discovering first dehydrocoupling catalyst for arsines. In these efforts we were further able to distinguish two separate mechanistic pathways of the dehydrocoupling catalysis. One pathway appeared to involve sigma-bond metathesis steps, analogous to our phosphine dehydrocoupling, while the other path gave evidence for a-arsinidene elimination. This work resulted in a publication in Dalton Transactions.
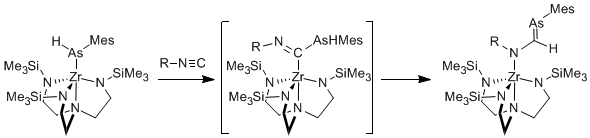
We have further discovered that these zirconium arsenido complexes readily react with isocyanides to give the 1,1-insertion products. When a primary arsenido ligand is used, the insertion product is not thermally stable, and rearranges to a complex featuring an arsaalkene moiety (Scheme). This transformation is general based on the range of aryl and alkyl substituents that are tolerated on the isocyanide. This is a highly efficient synthesis of arsaalkenes, which are normally prepared via condensation of dehydrohalogenation reactions.
Additionally, over the last year of the award, we have investigated some of the fundamental reactivity of our key precursor. We discovered that a cyclo-metalated derivative, [k5-N,N,N,N,C-(Me3SiNCH2CH2)2NCH2CH2NSiMe2CH2]Zr (1) is readily prepared by reaction of Zr(CH2Ph)4 with N(CH2CH2NHSiMe3)3. Complex 1 is a general precursor to complexes featuring ZrE bonds, where E can be carbon, phosphorus, nitrogen, oxygen, sulfur, or arsenic. Thus, complex 1 is incredibly useful in the preparation of catalysts and model complexes for study.
Complex 1 exhibits reaction chemistry as well. We have demonstrated a
number of insertion reactions in to the ZrC bond including 1,1-,
1,2-, and 1,3-insertions with isocyanides, ketones,
and organic azides as representative substrates, respectively. In a collaboration with Prof. Paul Hayes of the
Additional study of complex 1 suggests that bond formation with EH bonds is a concerted process, potentially proceeding by sigma-bond metathesis. Experiments with samples of para-enriched hydrogen showed a significant enhancement in the 1H NMR spectrum, consistent with this conclusion. We are completing kinetic isotope experiments, which will bring some additional insight regarding the mechanism of EH bond activation.
A final area that we have pursued with PRF support is the hydrophosphination of unsaturated substrates. In these reactions it was found that (N3N)ZrPPh2 is a catalyst for the addition of diphenylphosphine to terminal alkynes and carbodiimides. In the hydrophosphination of alkynes, vinyl phosphines are formed in good yields, but the reaction is sluggish. Further investigation revealed the formation of alkynyl complexes that act to inhibit the catalysis. For the carbodiimide substrates, the reactions were considerably more facile, suggestive of no substrate inhibition. Competition experiments and stoichiometric insertion reactions with polar unsaturated organic small molecules suggest that the reaction proceeds via insertion of the substrate into the ZrP bond. Our complexes represent a rare example of an early transition-metal hydrophospination catalyst, and the only example of an early metal catalyst that undergoes hydrophosphination via insertion of the unsaturated substrate.
Overall, PRF support has allowed us to significantly advance bond-forming reactions and catalysis featuring triamidoamine-supported zirconium complexes.



