Reports: AC3
47214-AC3 Oxygen Activation by Isopenicillin N Synthase - The Yin and Yang of C-H Activation
During year 1, we trapped and characterized an intermediate in the reaction of the IPNS:Fe(II):ACV complex with O2. Its parameters suggested that it is the second C-H-cleaving complex of the proposed mechanism, which is a Fe(IV)-oxo complex. During the second year, more insight into the mechanism of IPNS has emerged from additional studies carried out during this funding period. From these studies, which are described in the following, Scheme 1 was obtained. We intend to publish these results in the Proceedings of the National Academy of Sciences in the near future.
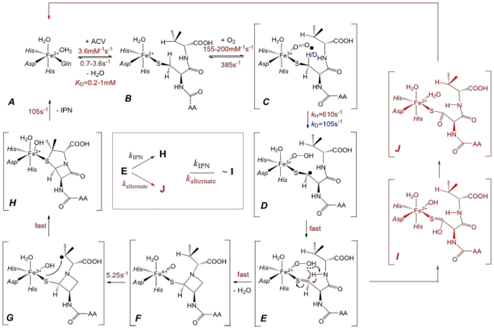
Scheme 1
Trapping of the proposed Fe(III)-superoxo intermediate (C in Scheme 1) by using A[3,3-d2-C]V, selectively deuterated at the target Cβ-H bond of cysteine.
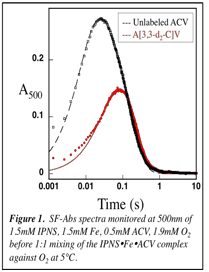
In the presence of unlabeled natural substrate, we obtained evidence for the second C-H-cleaving intermediate (F), but no evidence for accumulation of the first C-H-cleaving intermediate (C). We proposed that use of the specifically labeled substrate A[3,3-d2-C]V might slow decay of C and allow for its accumulation. We purchased A[3,3-d2-C]V and carried out stopped-flow absorption (SF-Abs) and freeze-quench Mssbauer (FQ-Moss) experiments. Monitoring the reaction between IPNS:Fe(II):A[3,3-d2-C]V and O2 by SF-Abs shows that F, which absorbs at 500 nm, forms more slowly, which is consistent with our expectation that a step prior to F formation is slowed down (Figure 1). Also consistent with this assignment, the decay of F is not perturbed by the isotopic substitution.
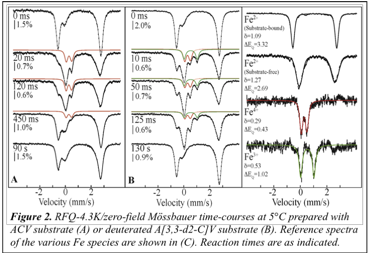
Direct evidence for accumulation of a new intermediate, presumably the
C-H-cleaving intermediate C, was
obtained by FQ-Moss experiments (Figure 2, middle panel). A new quadrupole
doublet (green) accumulates at early reaction times, before it decays to the
previously studied Fe(IV)-oxo complex, F
(red). For comparison, we also show the same experiment carried out with
unlabeled substrate (left panel). Deconvolution of the spectra into the various
subspectra (right panel) allows the parameters of the new intermediate to be
 determined: d
= 0.53 mm/s and ΔEQ = 1.02 mm/s. The isomer shift of 0.53 mm/s
is typical of high-spin Fe(III), and the quadrupole doublet feature indicates
that the high-spin Fe(III) site is associated with an integer spin ground
state. These findings are fully consistent with structure C of the mechanism, a Fe(III)-superoxo complex (SFe = 5/2 and Ssuperoxide = ½ couple
to a total spin of S = 2 or S = 3).
determined: d
= 0.53 mm/s and ΔEQ = 1.02 mm/s. The isomer shift of 0.53 mm/s
is typical of high-spin Fe(III), and the quadrupole doublet feature indicates
that the high-spin Fe(III) site is associated with an integer spin ground
state. These findings are fully consistent with structure C of the mechanism, a Fe(III)-superoxo complex (SFe = 5/2 and Ssuperoxide = ½ couple
to a total spin of S = 2 or S = 3).
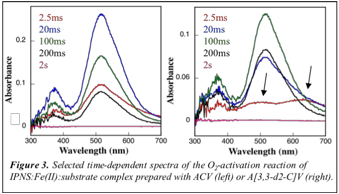
We have also more carefully studied the wavelength-dependent SF-Abs spectra of selected early reaction time points (Figure 3), in order to see whether the intermediate exhibits absorption features. We have evidence for new absorption features at ~600 nm and ~480 m (see arrows), which are only detected in the reaction with A[3,3-d2-C]V .
Evidence for a second reaction pathway.
 Using
the intense absorption band at 500 nm of the Fe(IV)-oxo (F), we carried out numerous SF-Abs studies in varying conditions
(many, but not all of these experiments were summarized in the previous
progress report). These studies have clearly shown that the reaction between
the IPNS:Fe(II):ACV complex and O2 is first order in each reactant.
From the rate constant determined from these simulations, we concluded that the
Fe(IV)-oxo complex should accumulate to >80%. However, by FQ-Moss, which
allows us to determine the absolute amount of the Fe(IV)-oxo complex, we only
detect ~50%. In order to resolve this discrepancy, we carried out SF-Abs and
FQ-Moss experiments under identical conditions. The results indicate that the
reaction kinetcs determined by the two methods correlate well. From these
studies it is possible to calculate the extinction coefficient (ε500
= 2.7 mM-1cm-1) for the Fe(IV)-oxo complex and
determine its absolute amount accurately by SF-Abs. Yet, the comparison between
the two methods did not resolve the low amount of Fe(IV)-oxo detected by
FQ-Moss.
Using
the intense absorption band at 500 nm of the Fe(IV)-oxo (F), we carried out numerous SF-Abs studies in varying conditions
(many, but not all of these experiments were summarized in the previous
progress report). These studies have clearly shown that the reaction between
the IPNS:Fe(II):ACV complex and O2 is first order in each reactant.
From the rate constant determined from these simulations, we concluded that the
Fe(IV)-oxo complex should accumulate to >80%. However, by FQ-Moss, which
allows us to determine the absolute amount of the Fe(IV)-oxo complex, we only
detect ~50%. In order to resolve this discrepancy, we carried out SF-Abs and
FQ-Moss experiments under identical conditions. The results indicate that the
reaction kinetcs determined by the two methods correlate well. From these
studies it is possible to calculate the extinction coefficient (ε500
= 2.7 mM-1cm-1) for the Fe(IV)-oxo complex and
determine its absolute amount accurately by SF-Abs. Yet, the comparison between
the two methods did not resolve the low amount of Fe(IV)-oxo detected by
FQ-Moss.
This conundrum can be solved by invoking a branchpoint in the reaction
cycle between oxygen addition (i.e. B
C in Scheme 1) and generation of the Fe(IV)-oxo complex. We propose
that the reaction cycle bifurcates at state E. In the natural reaction, E
decays by formation of the Fe(IV)-oxo complex. This reaction step also involves
formation of the Fe(IV)-oxo complex. This reaction step also involves formation
of the β-lactam
ring, which according to computational studies by
 Morokuma and co-workers may
have a large activation barrier.1 An alternative decay pathway for E
may involve formation of a thiocarboxylate product (J in Scheme 1). This product has been experimentally observed by
Baldwin and co-workers with a substrate analogue, in which the valine amide is
replaced by an ester group.2 A conceivable reaction path to this
product may involve attack of the distal O-atom of the hydroperoxide ligand in E on the thiocarbonyl group to yield
the thiocarboxylate (J).
Dissociation of the alternative product would regenerate the IPNS:Fe(II)
resting state (A) and feed back into the catalytic cycle.
Morokuma and co-workers may
have a large activation barrier.1 An alternative decay pathway for E
may involve formation of a thiocarboxylate product (J in Scheme 1). This product has been experimentally observed by
Baldwin and co-workers with a substrate analogue, in which the valine amide is
replaced by an ester group.2 A conceivable reaction path to this
product may involve attack of the distal O-atom of the hydroperoxide ligand in E on the thiocarbonyl group to yield
the thiocarboxylate (J).
Dissociation of the alternative product would regenerate the IPNS:Fe(II)
resting state (A) and feed back into the catalytic cycle.
This model is attractive, because it allows for all experimental observations to be rationalized. We are now carrying out experiments to test this hypothesis. We are using liquid chromatography coupled to mass spectrometry (LC/MS) to detect the proposed thiocarboxylate. We have identified a peak with the correct m/z ratio for the alternative thiocarboxylate product and will carry out additional control experiments (i.e. carry out the reaction with 18O2 to see a shift of the peak tentatively assigned to the thiocarboxylate by +2 m/z units).
Additionally, we have begun collaborating with Frank Neese's group from the University of Bonn, Germany, to correlate the experimentally observed spectroscopic properties of the two reaction intermediates to those calculated for hypothetical model structures.
Literature Cited:
1. Lundberg, M.; Siegahn, P. E. M.; Morokuma, K. "The Mechanism for Isopenicillin N Synthase from Density-Functional Modeling Highlights the Similarities with Other Enzymes in the 2-His-1-Carboxylate Family" Biochemistry 2008, 47, 1031-1042.
2. Daruzzaman, A.; Clifton, I. J.; Adlington, R. M.; Baldwin, J. E.; Rutledge, P. J. "Unexpected Oxidation of a Depsipeptide Substrate Analogue in Crystalline Isopenicillin N Synthase" ChemBioChem 2006, 7, 351-358.



