Reports: GB4
47864-GB4 Generation and Kinetic Studies of High-Valent Metal-Oxo Intermediates
Specific project objectives
Objective I: Chemical and photochemical generation of highly reactive porphyrin- ruthenium(V)-oxo species
Objective II: Comprehensive kinetic studies and mechanistic investigation of high-valent metal-oxo species
Scientific and Technical Description of the Results
Chemical generation of ruthenium(V)-oxo porphyrin species
As proposed, we tried to produce and detect the ruthenium(V)-oxo species by oxidation of the porphyrin ruthenium(III) precursors with two-electron sacrificial oxidants. Thus, we followed the reported procedure to synthesize the RuIII(TPP)(OEt), confirmed by the known UV-vis absorption and by a typical EPR signal of a ruthenium(III) species. However, treatment of the ruthenium(III) precursor by the sacrificial oxidants, including peroxyacetic acid, m-CPBA, iodosylbenzene and 2,6-dichlorpyridine, does not result in any change of UV-vis absorption, and no detectable intermediate was observed even at low temperature (-40 oC). These results suggested that the chemical oxidation of ruthenium(III) is not viable to generate the detectable ruthenium(V)-oxo species presumably due to its high reactivity and low concentration.
Photo-induced ligand cleavage reactions
During the current period, we have studied the photo-induced ligand cleavage reactions to generate the putative ruthenium(V)-oxo species. Firstly, we tested the photo-labile precursor 2, which was prepared by the reaction of 1 with an excess of pyridine-N-oxide (Scheme 1 and Figure 1A). Irradiation of 2 with 355 nm laser light in CH2Cl2 solution gave a transient that has a Soret band absorbance at ca. 395 nm. When excess cyclohexene was present, the decay of the formed species accelerated linearly with the substrate concentration (Figure 1C). The plot slope gave a secondary rate constant (k2) of 740 M-1 s-1, which is 4-5 orders of magnitude greater than that of reaction of the well characterized trans-dioxoruthenium(VI) complex. The generated transient species was tentatively assigned the ruthenium(V)-oxo structure 3 based on its unique UV-visible spectrum and its very high reactivity. With 2,6-dichloropyridine N-oxide for precursor, we obtained similar result.
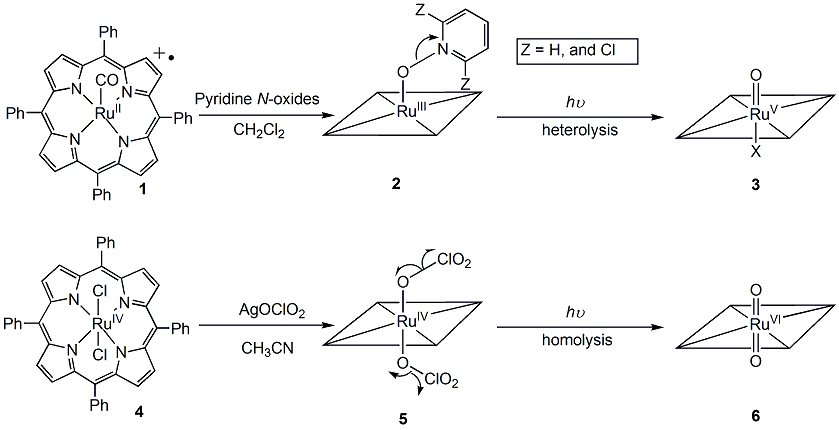
Scheme 1: Photo-induced ligand cleavage reactions
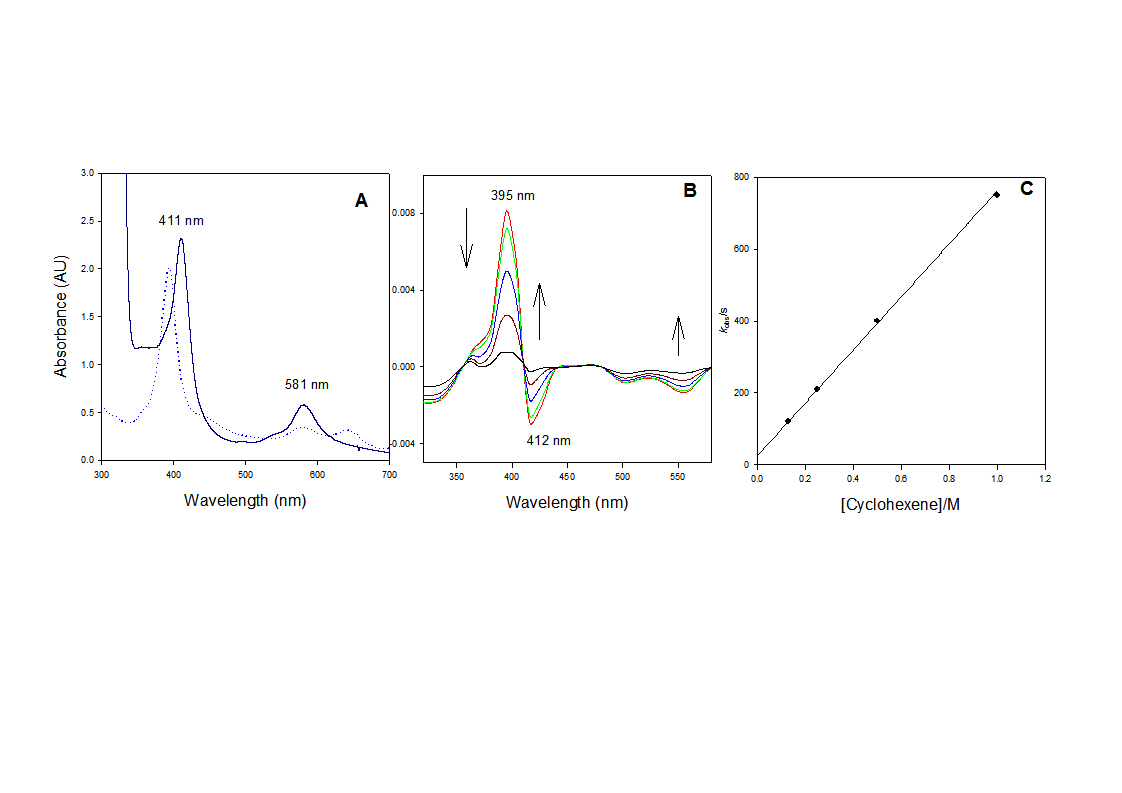
Figure 1: (A) UV-visible spectra of 2 (solid lines) and 1 (dotted lines) in CH2Cl2. (B) Time-resolved spectrum for 100 ms following 355 nm irradiation of species 2 in CH2Cl2 at 25 oC. (C) Observed rate constants for reactions with cyclohexene in CH2Cl2 at 25 oC, monitored at 395 nm.
Secondly, we also investigated the photolysis of porphyrin-ruthenium(IV) chlorate (5) which typically undergoes homolytic cleavage of the O-Cl. The precursor was prepared from the dichlororuthenium(IV) complex 4 by exchanging of the counterion with Ag(ClO3) in CH3CN. However, irradiation of complex 5 with 355 nm laser light gave a relatively stable species with a Soret band of lmax at 418 nm, which was characteristic absorption for the well-known trans-dioxoruthenium(VI) porphyrin complex, i.e. RuVI(O)2TPP (Scheme 1).
Photo-disproportionation of a diruthenium(IV)-m-oxo bisporphyrin complexes
In literature, photo-disproportionation of the m-oxo iron(III) dimer was well established to form iron(II) and porphyrin-iron(IV)-oxo species concomitantly. We suggest that the photo-disproportionation of diruthenium(IV)- m-oxo bisporphyrins may also produce the terminal ruthenium(V)-oxo intermediate and ruthenium(III) porphyrin (Eq. 1):
![]() (1)
(1)
To test above hypothesis, the diruthenium(IV)- m-oxo bis[5,10,15,20-tetraphenylporphyrin(TPP)] precursor (7) was synthesized. The initial hydroxyl axial ligand could be replaced with chloride to form [RuIV(TPP)Cl]2O (Figure 2A). At ambient temperature, irradiation of the complex [RuIV(TPP)OH]2O with 355 nm laser light in CH3CN solution produced a shot-lived transient (Figure 2B), very similar to that formed in photo-induced reaction, as determined by UV-vis spectra and kinetic behavior. The species decayed over 100 ms to form a species at 410 nm, known for RuIII species (Figure 2B). The k2 of 1800 M-1 s-1 with cyclohexene was obtained in CH3CN (Figure 1C). When the photo-disproportionation reaction was carried out in CH2Cl2, a second rate constant (k2 = 670 M-1 s-1) was obtained, confirming that the same oxidizing species was produced from both methods.
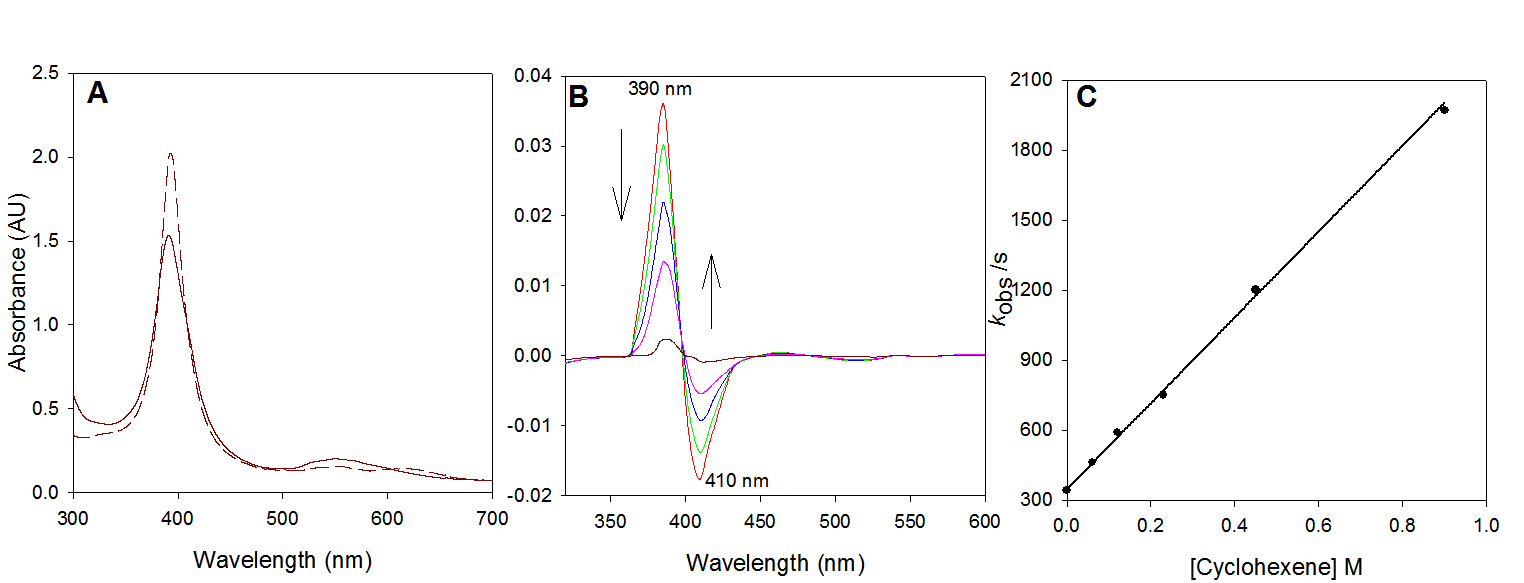
Figure 2: (A) UV-visible spectra of [RuIV(TPP)OH]2O (solid lines) and [RuIV(TPP)Cl]2O (dotted lines) in CH3CN. (B) Time-resolved difference spectrum over 100 ms following 355 nm irradiation of [RuIV(TPP)OH]2O in CH3CN at 25 oC. (C) Observed rate constants for reactions with cyclohexene in CH3CN at 25 oC, monitored at 390 nm.
Photo-disproportionation of a diiron(IV)-m-oxo biscorrole complexes
We extended the scope of the photo-disproportionation reactions with the utilization of iron corroles. In our published results, we demonstrated that iron(IV) m-oxo corrole dimers could form a reactive iron(V)-oxo intermediate, and potentially to provide another aerobic catalytic protocol as well. The bis-corrole diiron(IV)-oxo complex 8 was prepared and characterized by UV-visible, 1H NMR, and 19F NMR spectra that matched those previously reported in 2001 by Simkhovich et al. The UV-visible spectra of complex 8 and its corrole-iron(III) precursor are shown in Figure 3A.
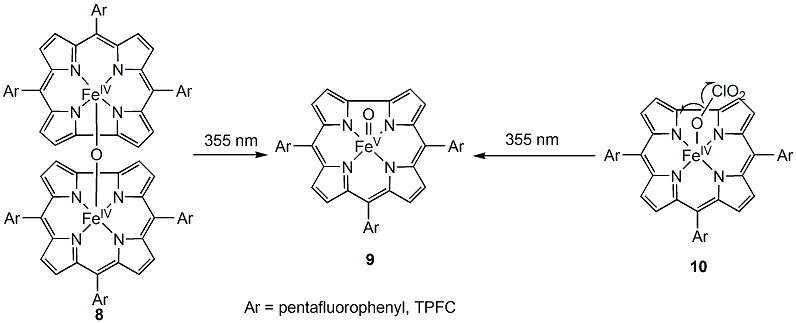
Scheme 2: Formation of an iron(V) corrole species by photolysis of either m-oxo iron(IV) biscorrole dimer (8) or a iron(IV) chlorate (10).
Irradiation of complex [FeIV(TPFP)]2O with 355 nm laser light in CH3CN solution produced a highly reactive transient with a Soret band absorbance with lmax at 415 nm (Figure 3B), the same as that of the species formed by photo-induced ligand cleavage reaction, reported by Newcomb and coworkers in 2005. The observed rate constant for cyclooctene is 5.6 ± 103 M-1 s-1, again, consistent with the previously reported one. Kinetic results with other substrates confirmed that same oxidizing species was produced from both routes.
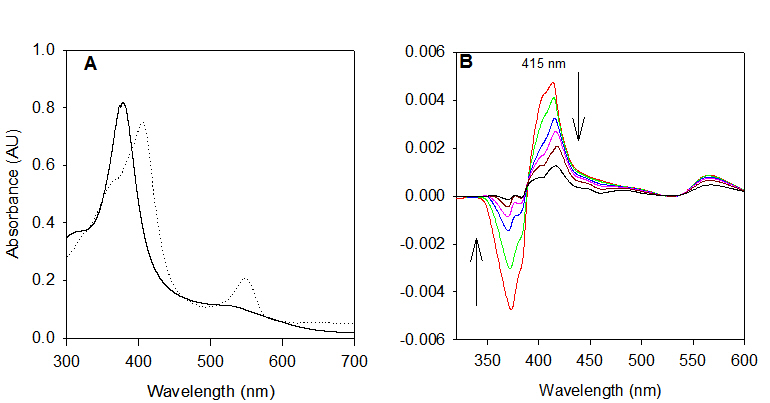
Figure 3: (A) UV-visible spectra of [FeIV(TPFC)]2O (solid lines) and FeIII(TPFC)(OEt)2 (dotted lines) in CH3CN. (B) Time-resolved difference spectrum over 10 ms following 355 nm irradiation of [FeIV(TPFC)]2O in CH3CN at 25 oC.
The potential for a green catalytic system was evaluated in the aerobic oxidation of cis-cyclooctene (Scheme 3). The reaction system consisted of 1 mmol of 8 in 5 mL of acetonitrile containing 5 mmol of cis-cyclooctene. Dry air was bubbled through the solution as it was irradiated with 400-500 nm light. After 48 hours of photolysis, cis-cyclooctene oxide was obtained as the only identifiable oxidation product (>95% by GC) with ca. 200 turnovers of catalyst.
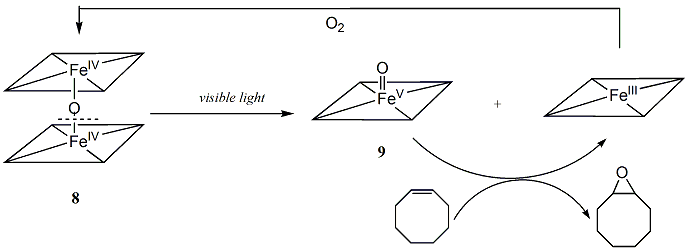
Scheme 3: Catalytic oxidation by the diiron(IV) m-oxo biscorroles using visible light and O2



