Reports: B4
47629-B4 Pentacyclo[4.3.0.0(2,4).0(3,8).0(5,7)]non-4-ene: Synthesis, Reactivity, Matrix Isolation Spectroscopy, Calculations, and Physical Study of Reaction Products
Previously, undergraduate students in my research group have shown that treatment of diiodide 1 with n-butyllithium (or t-butyllithium) at 0 oC in the presence of the trapping agent 1,3-diphenylisobenzofuran (DPIBF) furnishes a 25-30% yield of the "trapped" product 3, thus providing strong evidence for the formation of 2.
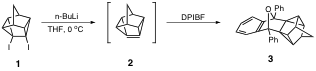
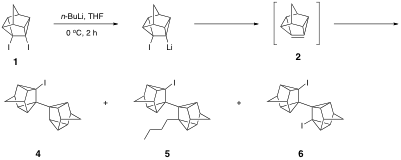
Dehalogenation of diiodide 1 with alkyllithiums in the absence of a trapping agent affords alkyllithium addition products 4,5, and 6 (and trace amounts of other alkyllithium addition products). These three products were also present in the aforementioned Diels-Alder trapping reaction of 1 with n-butyllithium or t-butyllithium. It is well known that alkyllithiums add readily to highly pyramidalized alkenes (see Tetrahedron 2005, 61, 5147). In order to further study the chemistry and reactivity of 2, an alternative route that does not utilize alkyllithiums to generate the pyramidalized double bond must be developed. The focus of our research during the grant period has been the synthesis of these alternative precursors.
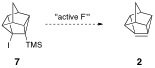
In his synthesis of cubene, Eaton observed that alkyllithiums present in the reaction mixture reacted rapidly with the pyramidalized double bond (see J. Am. Chem. Soc. 1988, 110, 7230-7232). The rapidity with which cubene reacted with alkyl lithiums prevented an exploration of the reactivity of the cubene double bond with reagents other than alkyllithiums, a situation similar to what is observed in our study. To combat this, Eaton showed that cubene could be generated in the absence of alkyl lithiums via the fluoride ion induced elimination of 1-halo-2-(trimethylsilyl)cubanes. In a similar manner, 4-iodo-5-(trimethylsilyl)pentacyclo[4.3.0.02,4.03,8.05,7]nonane (7) offers an alternative route to 2 via treatment with active fluoride.
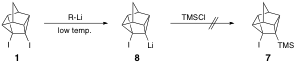
Previously, attempts to synthesize 7 via lithiation of diiodide 1 at low temperature followed by addition of chlorotrimethylsilane (TMS-Cl), were unsuccessful, as lithium iodide elimination from 8 was rapid even at low temperature.
Moreover, attempted lithiation of molecules of type 9 (E=-CO2CH3 ,-CO2H, -CO2C(CH3)3, -CN), and subsequent trapping with chlorotrimethylsilane also did not lead to molecules of the type 11.
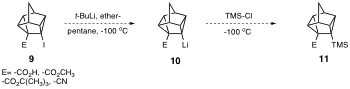
The synthesis of one or more of the derivatives of 11 would have allowed the successful synthesis of 7 via standard FGI (i.e. conversion of E to iodine via Barton iododecarboxylation). Indeed, we have already shown that Barton halodecarboxylation is a successful method for the introduction of iodine at this position of the ring system.
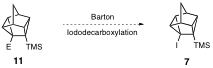
During the grant period my research group also synthesized iodoalcohol 12. We expected that in the treatment of iodoalcohol 12 with 2 plus equivalents of an alkyllithium, the first alkyllithium equivalent would deprontonate the hydroxyl group and the second equivalent would undergo lithium-iodine exchange to give 13, which would furnish 15 upon quenching with excess chlorotrimethylsilane. However, we learned via trapping studies that lithium-iodine exchange on 12 occurs prior to deprotonation, and the resultant alkyllithium then quenches itself to give 14.
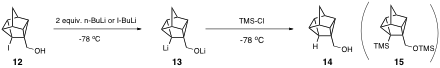
To combat this problem, my group protected the hydroxyl group of 12 as its trimethylsilyl ether 16 and then carried out the lithium-iodine exchange reaction with alkyllithiums. Preliminary results suggest that we have synthesized 18. At this point, we are not sure if 18 results from direct quenching of 17 with chlorotrimethylsilane, or if 17 undergoes retro-Brook rearrangement prior to quenching. Thus far, we have been unable to purify crude samples of what we think is 18, and thus that is the focus of our continuing research efforts in this area.
As an alternative approach to 7, we have previously shown that treatment of iodoester 19 with isopropylmagnesium chloride leads to cyclopropylmagnesium reagent 20. Quenching 20 with either methanol or benzaldehyde led to good yields of 21 and 22, respectively. However, quenching the reaction mixture with trimethylsilyl chloride did not lead to 23, despite warming and prolonged reaction time; only monoester 21 was formed upon workup.
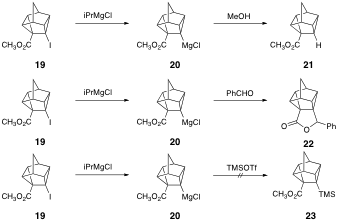
The reactions of Grignard reagents with triorganosilyl chlorides are known to be very sluggish and give satisfactory yields of only the simplest tetraorganosilanes. We surmised that a more highly reactive silylating agent would be required for this reaction, and thus attempted quenching the reaction mixture with trimethylsilyl triflate, a highly reactive silylating agent. The reaction yields of 23 are low, and thus far we have not been able to optimize the reaction to afford higher yields of 23. We are still hopeful that optimization of this reaction followed by Barton iododecarboxylation should lead to of 7. We will also continue to pursue the use of other reactive silylating agents for this reaction.



