Reports: AC3
46982-AC3 New Robust Tridentate N-Heterocyclic Carbene Ligands for C-H Activation
One of the most exciting advances in transition-metal catalyzed processes has been the move to use N-heterocyclic carbenes (NHCs) as ancillary ligands. This has been driven by a number of factors, which include the enhanced stability of NHC-containing complexes, particularly versus similar phosphine-based systems, and the enhanced reactivity of NHC-derived catalysts. However, as spectator ligands, NHCs have shown some potential drawbacks as the carbene moiety can participate in migratory insertion, reductive elimination, and "wrong-way" binding via C-H bond activation. Such non-innocent reactivities are likely sources of NHC-based catalyst deactivation.
Incorporation of NHCs into chelating arrays has been shown to generate particularly robust catalyst systems. For example, a palladium complex of a tridentate [CNC] system, which has pyridine flanked by two sterically unencumbered NHCs, has been reported to be stable to 180ûC in air and active for Heck catalysis. Interestingly, stoichiometric reactions done independently show that this type of tridentate NHC system can behave non-innocently by migratory insertion or reductive elimination behavior. In both cases, the NHC is positioned cis to the Pd-methyl unit, which is optimal for both of these kinds of processes.
Other tridentate ligand systems that incorporate NHCs are known, and those with the NHC positioned as the central donor should be less susceptible to these kinds of deleterious processes. In this paper, we document a C-C bond-forming process to generate a nickel(0) derivative that apparently involves the N-heterocyclic carbene moiety and the trans disposed hydride ligand. Mechanistic and computational studies show that this process is unlikely to involve square planar Ni(II) intermediates.
We have recently reported the preparation of a rigid tridentate PCP ligand with a saturated NHC moiety flanked by two o-phosphinoaryl substituents and have examined its coordination chemistry with a variety of transition metals, including nickel. Reaction of the imidazolidinium salt ([PCP]H)PF6 1 (where [PCP] is o-Pri2PC6H4(NC3H4N)o-C6H4PPri2) with Ni(COD)2 results in the formation of the nickel(II) hydride complex ([PCP]NiH)PF6 2, which has been characterized by both NMR spectroscopy and X-ray crystallography.
In the process of examining the reactivity of the nickel hydride derivative, we added excess ethylene to a solution of 2 in THF and observed a rapid color change from yellow to orange-red. Our expectation was the formation of a square-planar Ni(II) ethyl complex, ([PCP]NiEt)PF6 (3), via migratory insertion of ethylene into the Ni-H bond. Instead, the resultant product is formally a Ni(0) h2-iminium diphosphine complex, shown as 4 in Scheme 1, formed in 70% isolated yield. That this was not the simple Ni(II)-ethyl species 3 was evident from the 1H NMR spectrum, which showed rather broad signals, and in particular, a set of two broad peaks due to inequivalent methylene protons in the saturated carbene backbone, as well resonances due to as inequivalent isopropyl substituents.
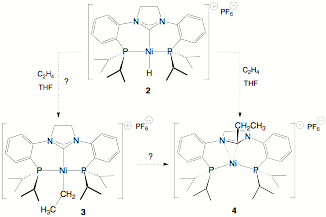
Scheme 1. The reaction of ethylene with the nickel hydride complex produces 4; how the nickel ethyl complex 3 is involved in this transformation is discussed below.
Single crystals obtained from THF were analyzed by X-ray diffraction; the solid-state molecular structure is shown in Figure 2, along with selected bond lengths and bond angles. What is immediately apparent is that C-C bond formation has occurred wherein an ethyl moiety is attached to C1, formerly the carbene carbon, and this ethylimidazolidinium unit is now p-bound to the formally Ni(0) center. In the solid state, this h2-C=N interaction renders the two phosphine donors inequivalent as well as the top and bottom of the p-bound heterocycle. Interestingly, in solution the 31P{1H} NMR spectrum of 4 shows a singlet for the ligated phosphines even down to -80ûC, which suggests that fast exchange of the h2-C=N unit between N1 and N2 is occurring on the NMR time scale. While this process equilibrates the two phosphine arms, it does not exchange the two isopropyl groups on each phosphorus as they remain inequivalent even at higher temperatures due to the different faces of the coordinated heterocycle above and below the P1-Ni-P2 plane.
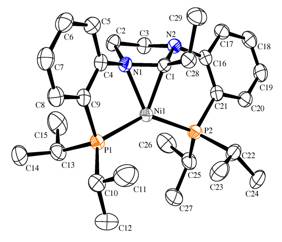
Figure 2. ORTEP diagram of the cation of 4. Selected bond lengths () and bond angles (û): Ni1-P1, 2.2295(8); Ni1-P2, 2.1424(8); Ni1-N1, 1.967(2); Ni1-C1, 1.899(3); N1-C1, 1.436(3); P1-Ni1-P2, 137.33(3); N1-Ni1-C1, 43.54(10); N1-C1-N2; 107.3(2); P1-Ni1-N1, 89.68(7); C1-Ni1-P2, 92.35(8); C1-C28-C29, 115.3(2).
Based on literature precedent, we assumed that a likely intermediate would be the square-planar nickel ethyl complex 3, formed via migratory insertion of added ethylene into the nickel hydride bond, which could undergo a C-C coupling process to generate the observed Ni(0) product 4. Another possible mechanism, but with no precedent to our knowledge, is the direct insertion of ethylene into the nickel-carbene bond followed by a C-H reductive elimination would also generate 4. The obvious problem with both proposed mechanisms is that reductive elimination processes normally require that the two ligands undergoing the rearrangement be cis-disposed, but in each process, the square planar Ni(II) center requires that the two ligands be trans disposed. Even in the presence of excess ethylene, any putative five-coordinate intermediate cannot rearrange to allow trans to cis isomerization of the migrating/eliminating ligands.
We were able to shed light on this process by collaborating with Professor Jennifer Green at Oxford University. Her DFT analysis showed that the steric protection provided by the trans-disposed di-isopropylphosphine groups made insertion of C2H4 occur via an unprecedented mechanism, that of apical formation of an agostic ethyl group, which rearranges to the observed Ni(0) complex via cis migration. This process obviates any requirement for a trans reductive elimination process.
This work has recently been published:
Steinke, T; Shaw, B. K.; Jong, H.; Patrick, B. O.; Fryzuk, M. D.; Green, J. C. "Non-Innocent Behavior of Ancillary Ligands. Apparent Trans Coupling of a Saturated N-Heterocyclic Carbene Unit with an Ethyl Ligand Mediated by Nickel", J. Am. Chem. Soc. 2009, 131, 10461-10466.



