Reports: G1
46202-G1 Catalytic Asymmetric Ketone Homologation. Development of Chiral Aluminum Complexes for the Formal Deoxygenative Insertion of Carbonyl Compounds into Acyclic and Cyclic Ketones
In year one of the funding period, my group discovered efficient catalysis of the diazoalkane–carbonyl reaction by scandium3+ salts. After a study focused on cycloalkanone electrophiles, aldehydes were tested.
 I. A General Approach to Acyclic Ketones by
I. A General Approach to Acyclic Ketones by
Carbon Insertion into the Formyl C-H Bond
Ketones are synthesized by a variety of means, one of the more common being a two-step process involving organometal addition to an aldehyde and oxidation. The aldehyde ¨ ketone conversion has also been accomplished in a single step by reaction with diazo compounds, yet limitations to the scope and efficiency of the reaction are unmet challenges. Given the broad efficacy of Sc3+ as a tunable Lewis acid for diazoalkyl carbon insertion with cyclic ketones, it appeared of value to extend this mild form of C–C bond formation to aldehyde acceptors.
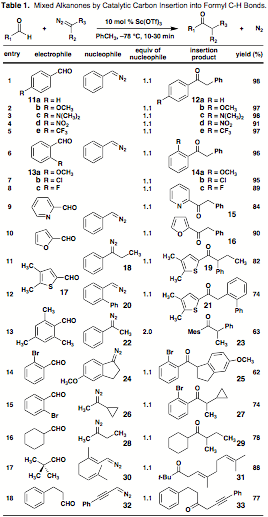
Convergency and a tolerance for steric bulk. A study began with the preparation of benzyl phenyl ketone (12a) from benzaldehyde and phenyldiazo-methane (entry 1 of Table 1). Dropwise addition of the nucleophile to a mixture of 11a and 10 mol % Sc(OTf)3 in toluene at –78 °C resembled a titration, with immediate dissipation of its signature red color and nitrogen evolution. After filtration through a pad of silica, 12a was isolated in 98% yield. There is no detectable union of the reactants in the absence of catalyst, even at 23 °C. Insertion events involving chosen electron-donating (OCH3 or N(CH3)2, entries 2 and 3) and -withdrawing (NO2 or CF3, entries 4, 5) groups situated para to the formyl carbon proceed smoothly in >90% yield. Such substituents can also be positioned ortho (96% yield for o-OCH3, entry 6), and entries 7 and 8 confirm that halogens are easily tolerated. The transformation is also successful for pyridine-2-carboxaldehyde (84% of 15, entry 9) and furfural (90% of 16, entry 10). Given the speed and efficiency of the process, efforts to lower the loading of catalyst were made. If reactions in entries 1, 3, 5 and 8 are performed with just 2 mol % Sc(OTf)3, the corresponding yields of pure product are 93, 89, 81, and 88%. Application to the synthesis of more elaborate (and chiral) ketones is also feasible. For example, thiophene carboxaldehyde 17 engages both an internal diazoalkane (18, entry 11) or one that is hindered on the basis of ortho substitution (20, entry 12). Steric crowding in the aldehyde is not a detriment either, as seen by the moderately effective coupling of mesitaldehyde with methyl phenyl diazomethane (22, 2 equiv, entry 13). Cyclic disubstituted diazomethanes can be employed as well; the one-step buildup of acyl indane 25 from electron-rich 24 and 2-bromobenzaldehyde is representative (entry 14). It was further of interest to explore mixed aryl-alkyl and alkyl-alkyl ketone preparations by the appropriate combination of aldehyde and diazoalkane. Merger of 2-bromobenzaldehyde with the potentially labile cyclopropyl methyl diazomethane (26) affords chiral aryl-alkyl product 27 in 74% yield (entry 15). Entry 16 shows that dialkyl ketone synthesis is viable for saturated internal diazo compounds and aliphatic electrophiles that are b-branched (78% of 29). Even pivaldehyde reacts in a coupling that is noteworthy since neither trans¨cis stereomutation of geranial-derived diazoalkane 30 nor conjugation of b,g-unsaturated product 31 is observed (entry 17). Alkynyl reactants are also suitable (77% of 33, entry 18).
II. Applying
the Aldehyde Insertion Reaction to the Total Synthesis of a Natural Product
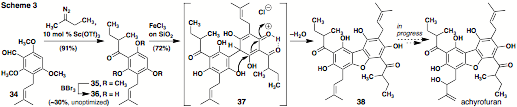
A five-step, biomimetic
synthesis of racemic 'pre-achyrofuran.' To test
the utility of catalytic carbon insertion with a complex aldehyde, we are
targeting achyrofuran, a potent South American medicinal agent with promise for
the treatment of type 2 diabetes (non-insulin-dependent diabetes mellitus, NIDDM). Arene
34 is available
in 80% yield on multigram scale by prenylation (n-BuLi, prenyl bromide) and
Vilsmeier-Haack formylation of 1,3,5-trimethoxybenzene. As shown in Scheme 3, its exposure to
2-diazobutane and 10 mol % Sc(OTf)3 under standard conditions
affords sec-butyl
ketone 35 with
high efficiency. After
demethylation, triphenol 36 undergoes oxidative dimerization with dibenzofuran ring
closure to give 38,
a likely biosynthetic precursor to the natural product. Regiocontrol in our access to
'pre-achyrofuran' derives from the facts that: (1) the indicated site of
Michael attack in 37 is doubly activated by virtue of the proximal acyl
substituent and (2) the phenol situated para to the electron-withdrawing group
is a better nucleophile than its counterpart three carbons away. Noteworthy is that the relative and
absolute configuration of the target is still unknown.
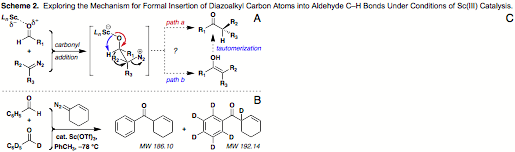
 Affirming the mechanism for
net C–H insertion through isotopic labeling. It is over a century since the first reactions of diazomethane with
aldehydes cleanly gave methyl ketones, and modern applications continue to be
reported. The general consensus is
that the reaction proceeds via concerted 1,2-migration of the C–H bond in the
diazonium betaine (Scheme 2A, path a), and its
status as premier migrating group is likely a result of its size. In the context of Sc-catalyzed reactions,
we have ruled out an alternative pathway involving SN2 closure to the epoxide and
subsequent rearrangement: exposure of either cis or trans stilbene oxide to 10 mol % Sc(OTf)3
in toluene at –78 °C affords diphenylacetaldehyde upon warming. Still, a third plausible mechanism
would invoke intermolecular E2 elimination of N2 to give an enol
intermediate followed by tautomerization (path b). Given our interest in asymmetric
catalysis of this transformation, we sought to unambiguously exclude the
possibility of an achiral reaction intermediate. To that end, the double-labeling experiment seen in Scheme
2B was performed. Sc-catalyzed
reaction of an equal mixture of benzaldehyde and perdeuteriobenzaldehyde with
3-diazocyclohex-1-ene provided only the products of intramolecular C–H
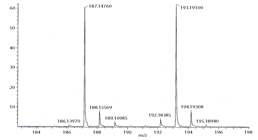
transfer according to high-resolution mass spectral analysis (ESI+). Although molecular ions corresponding
to deuterium crossover are detectable in the spectrum (Scheme 2C), control
experiments run independently with each starting material show that they occur
at levels consistent with natural abundance and the starting enrichment for d6-PhCHO (~97%). This conclusively validates
intramolecular C–H 1,2-shift as the operative mechanism, an outcome
fitting for enantioselective catalysis of carbon insertion with aldehyde
electrophiles.
Affirming the mechanism for
net C–H insertion through isotopic labeling. It is over a century since the first reactions of diazomethane with
aldehydes cleanly gave methyl ketones, and modern applications continue to be
reported. The general consensus is
that the reaction proceeds via concerted 1,2-migration of the C–H bond in the
diazonium betaine (Scheme 2A, path a), and its
status as premier migrating group is likely a result of its size. In the context of Sc-catalyzed reactions,
we have ruled out an alternative pathway involving SN2 closure to the epoxide and
subsequent rearrangement: exposure of either cis or trans stilbene oxide to 10 mol % Sc(OTf)3
in toluene at –78 °C affords diphenylacetaldehyde upon warming. Still, a third plausible mechanism
would invoke intermolecular E2 elimination of N2 to give an enol
intermediate followed by tautomerization (path b). Given our interest in asymmetric
catalysis of this transformation, we sought to unambiguously exclude the
possibility of an achiral reaction intermediate. To that end, the double-labeling experiment seen in Scheme
2B was performed. Sc-catalyzed
reaction of an equal mixture of benzaldehyde and perdeuteriobenzaldehyde with
3-diazocyclohex-1-ene provided only the products of intramolecular C–H
transfer according to high-resolution mass spectral analysis (ESI+). Although molecular ions corresponding
to deuterium crossover are detectable in the spectrum (Scheme 2C), control
experiments run independently with each starting material show that they occur
at levels consistent with natural abundance and the starting enrichment for d6-PhCHO (~97%). This conclusively validates
intramolecular C–H 1,2-shift as the operative mechanism, an outcome
fitting for enantioselective catalysis of carbon insertion with aldehyde
electrophiles.