Reports: AC6
48623-AC6 Multi-Scale Modeling of Reaction with Diffusion
Heterogeneous reactions carried out on nano-porous substrates are limited by both chemical kinetics and diffusion of reactants and products to and from catalyst sites. Fundamental understanding is difficult to achieve from experiment alone, since the measurements reflect the sum of many different effects diffusion to and from active sites, chemical kinetic limitations, selective adsorption of particular species, geometric constraints due to pore walls, surface defects, etc. The goals of this project are to achieve fundamental understanding of the influence of confinement in nano-pores on diffusion and chemical reaction, using theory and simulation at both the electronic and atomistic scales of matter.
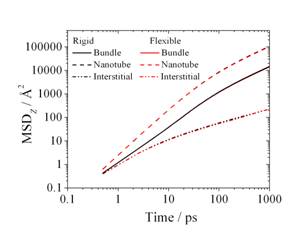
We have carried out molecular dynamics simulations for single walled carbon nanotubes having a wide range of pore diameters D and chiralities, and find that for pore diameters of 2.8s or greater, where s is the diameter of the diffusing molecule, the diffusion obeys Fick's Law, i.e. the molecules perform a three dimensional random walk, and the mean squared molecular displacement along the tube is proportional to the time, t. However, for tubes smaller than 2.8s the molecules can no longer pass each other and we observe single file diffusion, for which the mean squared displacement grows as the square root of time, i.e. much more slowly.
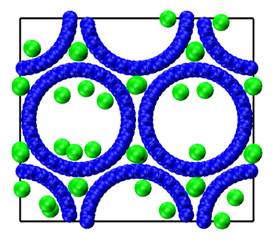
Many catalyst materials, as well as carbon electrodes used in fuel cells and supercapacitors, have a range of pore sizes that include both micropores (D<2 nm) and mesopores (2<D<50 nm), and both single file and Fickian diffusion are observed in different regions of the same material. We have used molecular dynamics simulations to study the diffusion of argon in two such model systems [1], a bundle of single walled (25,0) carbon nanotubes, and a widely used industrial activated carbon, BPL. In both cases we observe a bimodal diffusion mechanism with molecules exhibiting single file in small pores, and Fickian diffusion in the larger pores (Figure 1).
(a)
(b)
Figure 1: Diffusion of argon (green spheres) in a
bundle of (25,0) single walled carbon nanotubes for a bulk gas pressure of P/Po = 1.2 x
10-4 and a temperature of 120 K, where We
have also performed an ab initio density functional theory study
of the dissociation of water to generate hydrogen on titanium-decorated buckyballs, followed by storage of the generated hydrogen
[2]. We find that both single Ti atoms
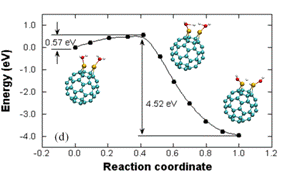
and Ti dimers on C60 (Figure 2 (a)) act as
catalyst sites for water dissociation with much lower energy barriers (~0.2-0.6
eV) than for water splitting in the bulk phase (~5 eV). Our
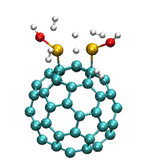
calculations show a high hydrogen storage of 5 hydrogen molecules for
Ti-dimer-C60 in the absence of water (Figure 2 (b)), with the
hydrogen molecules chemisorbed with an average binding energy of about -0.57 eV. However, when
water molecules are present the hydrogen molecules are only weakly bound by physisorption.
(a)
(b)
Figure 2. (a) Dissociation of two water molecules over a
Ti dimer bonded to C60, showing the
reaction pathway and energy barrier. (b)
H2 storage after two water molecule dissociation on the same
material. Here C is cyan, Ti is yellow,
O is red and H is white.