Reports: AC1
47257-AC1 Methods for Ligand Scaffold Optimization in Catalysis
This grant seeks to develop and exploit ligand scaffold optimization as a novel strategy for improving catalyst efficiency. The overall goal of this proposal is to develop methodologies which allow for scaffold optimization and use it to design, prepare, and evaluate focused libraries of new chiral ligands for use in catalytic asymmetric synthesis. The specific aims are to (i) carry out mechanistic studies to improve our understanding of the current generation of self-assembled ligands (SALs), (ii) evaluate new, complementary structural motifs and metal coordination geometries for the preparation self-assembled ligands and catalyst systems, (iii) evaluate the use of boracycles as an alternative motif for self-assembled ligands, and (iv) generate diverse ligand scaffold libraries via click chemistry.
Progress has been made on several of
the specific aims, particularly, specific aim iv.
Following up on a 2008 Organic Letters communication, a full paper comparing
the results obtained using two different ligating groups with a series
click-connected ligand scaffolds is in preparation. Publication is delayed
somewhat as we work to grow crystals suitable for structure determination. At
present the crystals are too small for structure determination. Computational studies
carried out by our collaborator, Prof. Libbie Pelter (
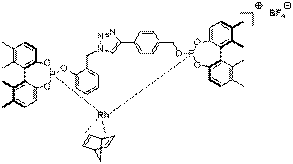
Figure 1. Structural studies on a successful Click-Connected catalyst are underway.
In addition, new studies carried out
by Mr. Nathan Thacker and Mr. Kaz Toyama, graduate
students in the UNL Chemistry departments, assisted by summer undergraduate research
student, Mr. Jeffrey O'Neill (
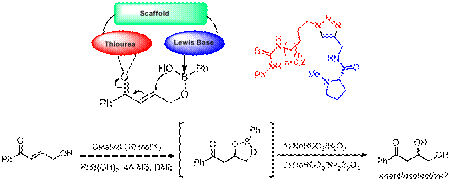
Figure 2. The design and first generation bifunctional dual activation catalysts for the oxy-Michael reaction exploiting the ability to generate diverse catalyst scaffold libraries for scaffold optimization.
In the proposal it was
noted that preliminary experiments had been carried out on the combination of
the chiral, homoleptic complex (SS,SS)-27 with the sterically encumbered tetramethyl-box derivative 25a (R1 = H) and the unsubstituted-box
derivative 25b (R2 = H).
Remarkably, 1H NMR analysis had indicated both combinations undergo
facile exchange with high selectivity for formation of the heteroleptic
complexes (i.e., 28a (R = Me) and 28b (R =
H). We had proposed to characterize and crystallize the new heteroleptic
complexes in order to better understand the origin of their large thermodynamic
preference. This past summer, an undergraduate, Ms. Kirsten Johnson from
neighboring
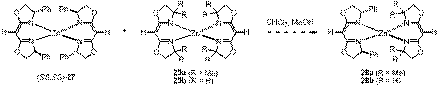
Figure 3. Alternative structural motifs that apparently also direct heteroleptic self-assembly are under investigation.



