Reports: AC4
46191-AC4 Electrochemical Studies on the Effect of Hydrogen Bonding and Proton Transfer Reactions of Organic Redox Couples
The goal of this grant is to develop a better understanding of the role of hydrogen bonding and proton transfer in organic redox chemistry by doing detailed cyclic voltammetry (CV) studies of organic redox couples in the presence of hydrogen donors and acceptors of various strength. One of the main systems proposed for investigation was oxidation of 2,3,5,6-tetramethylphenylenediamine, H2PD, in acetonitrile in the presence of different pyridines. The phenylenediamines are particularly interesting to study because their redox behavior shows obvious similarities to the more well-studied quinones, but there also important differences. For example, while the quinones start out neutral and become increasingly basic as they are reduced, the phenylenediamines start out basic and become increasingly acidic as they are oxidized. We believe that a comparison of the similarities and differences between the voltammetry of both compounds in conjunction with their chemical properties will yield useful insights into the redox chemistry of both couples.
While substantial experimental work has been done on this project, and it is our intent to complete the studies of H2PD with different bases, we have spent most of our effort in the past year on two related, but slightly different topics. The first is necessary for completion of the main project and that is to determine why the second CV wave of H2PD in acetonitrile is too small. The unexpected answer, detailed below and on the research nugget, reinforces the importance of H-bonding and proton transfer in these systems. The second project is a continuation of our investigation of the electrochemistry of phenylenediamines modified with an urea functionality in order to enhance H-bonding with other molecules.
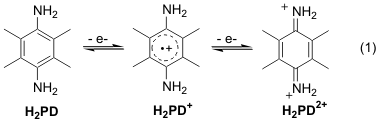
The too small second CV wave of H2PD. CV's of redox couples like quinones and phenylenediamines that undergo two successive electron transfer reactions in the absence of proton donors, eq 1, are expected to show two CV waves of approximate equal size for each electron transfer. However, with NH-containing phenylenediamines and quinones the second wave is often observed to be too small. Interestingly with N-Me phenylenediamines, which undergo similar redox reactions, the second wave is the correct height, suggesting that the too small CV wave might originate from either H-bonding or proton transfer involving the NH's. There are several bi-molecular reactions between the intermediates in solution that might explain this phenomenon. Some of these have been used to explain similar type of behavior with quinones. In order to test this with H2PD we looked at the concentration dependence of the CV's over a wide range, from 5mM to 5 mM. Since the rates of bi-molecular reactions will decrease with concentration faster than unimolecular reactions (such as electron transfer at the electrode), if a solution-phase bimolecular reaction was responsible for the too small second CV wave, then the wave should get larger relative to the first as the concentration decreases. In fact, the exact opposite is observed, with the second wave disappearing completely at the lowest concentrations. Since UV/Vis spectroelectrochemical data supports the expectation that the second CV wave is due to oxidation of solution-phase radical cation, H2PD+, and no CV wave means no radical cation, we are left with the entirely unexpected conclusion that the radical cation is not the initially formed product upon one electron oxidation of H2PD.
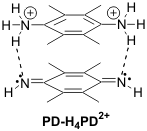
Instead, another product, which can not be further oxidized, is formed first, and at higher concentrations this kinetic product breaks apart to the thermodynamically stable radical cation. Extensive investigations have lead us to propose that the initial product is the pi-dimer PD-H4PD2+ formed on the electrode surface via proton exchange and H-bonding between two radical cations.
Very strong redox-dependent H-bonding with a phenylenediamine urea derivative. In last years report we focused on our work with a NMe2-substituted diphenylurea derivative. Upon one electron oxidation, the strength of H-bonding to a diamide guest increases by >2000-fold in 0.1 M NBu4B(C6F5)4/CH2Cl2. This was by far the largest binding enhancement that had been reported to date for oxidation-based, redox-dependent H-bonding to a molecular guest. We have continued these studies with a bis-NMe2-substituted derivative that can be oxidized by two successive one electron steps. Even stronger binding enhancements, up to 100,000-fold, are observed in this case.



