Reports: AC7
42995-AC7 Interfacial Structure and Fluctuations in Block Copolymer Brushes: Tailoring Switchable Surfaces
Diblock copolymer brushes (DCBs) have garnered interest due to the observation that their surface character can be altered by treatment with a solvent selective for one of their blocks. Zhao1 et al. showed for the first time that the surface of a polystyrene-b-poly(methyl methacrylate) (PS-b-PMMA) brush can be rearranged by changing the character of the solvent contacting the brush before it is dried. The objectives of this project were to clarify the structures of DCB as-deposited and switched states and understand the fluctuations of the DCB interfaces. These are first steps to understanding and controlling surface rearrangement.
Neutron reflectometry(NR) measurements have revealed the internal structure of dPS-b-PMA and PMA-b-dPS DCBs and this year we have compared our NR measurements of interface widths (w) with theoretical results from a collaborator, Prof. David Wu of the Colorado School of Mines. The interface widths for brushes with a PMA block tethered to the substrate are smaller than for brushes with a dPS block tethered to the substrate. Experimental interface width values are consistent with expectations from self-consistent field theory for brushes with a dPS bottom block. A scaling theory identifies a crossover as (d/Rg)2 > χN from the classical Helfand-Tagami regime, where w ≈ b/√χ, to a new stretched interface regime where w ≈ d/(χN). Scattering data from DCBs exposed to various solvents were also analyzed this year in order to probe the changes in structure that come with "switching" in solvent.
Describing the rearrangement of a DCB also requires an understanding of fluctuations of both interfaces internal to the DCB as well as fluctuations of the interface with the DCB's surroundings. Therefore, we have investigated brush surface height fluctuations using x-ray photon correlation spectroscopy (XPCS). As previously reported, the data reveal that both polystyrene and poly(n-butyl acrylate) homopolymer brushes have structure at the surface with length scales in the region of 620 nm to 3100 nm, but the surface features show no relaxation in a time window of 0.1 seconds to 1000 seconds, even at temperatures more than 130 °C above the glass transition temperature of corresponding untethered chains. In recent work we have shown that even reduction of the grafting density from 0.6 to 0.1 chains/nm2 does not bring the relaxation of surface height fluctuations into the XPCS measurement window. This remarkable change in surface dynamics is more profound than that seen in ultrathin films of untethered chains and is a result of covalent tethering. The ability to alter surface dynamics by tethering could be used to control wetting and adhesion of surfaces.
As a first step toward determining the fluctuations of DCB interfaces with low molecular weight solvent we attempted to swell homopolymer PS brushes with low molecular weight (2.2k) PS chains to see how this alters the brush surface dynamics. In cases in which the amount of 2.2k chains placed in contact with the brush was more than could be accommodated by swelling the brush, we discovered a very interesting impact of the underlying brush on the dynamics of the upper layer of "untethered" chains. A sketch of this sort of sample is shown in the inset of Fig. 1. Fig. 1 further shows the experimentally measured relaxation time constants (τ) as a function of in-plane wave vector value, q, at 100 °C for the surface of a layer of 2.2k PS chains atop four sorts of substrates. The most rapid dynamics were seen for the case of the 45 nm thick 2.2k PS film deposited directly on a silicon wafer from which the native oxide has been etched using hydrofluoric acid. Also shown are relaxation times at the same temperature for three samples for each of which the PS film of 2.2k chains was spuncast onto a densely grafted (σ = 0.5-0.7 chains/nm2) brush using conditions such that after spinning the thickness of the overall sample (brush+free chains), as determined by X-ray reflectivity, was approximately 45 nm greater than the thickness of the brush itself. When such a film is on top of a 21 nm thick PS brush, the relaxation time of the outer surface is 2 times larger than that for the corresponding film on a silicon substrate. When the brush thickness increases to 44 nm and 76 nm, the relaxation becomes 5 times and 14 times slower than that of a film on bare silicon substrate, respectively. NR measurements show that the interpenetration between the layers determines the relaxation time of the surface by altering the untethered chain layer that does not penetrate into the brush layer.
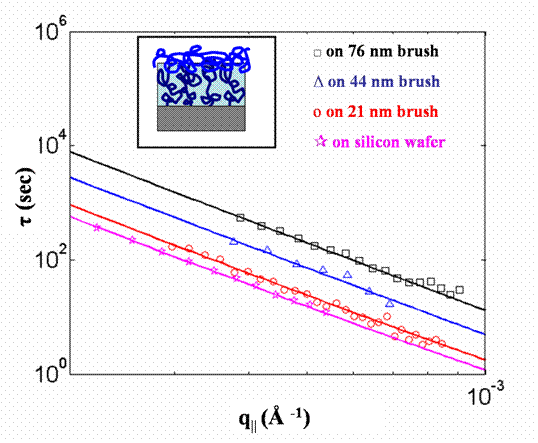
Figure 1. Time constants (τ) at 100 °C for 45 nm film of untethered PS chains deposited on Si wafer (stars), and brushes with σ = 0.6 chains/nm2 and thicknesses of 21 nm (circles), 44 nm (triangles) and 76 nm (squares). Curves represent least-squares fits based on over-damped capillary wave theory for a single viscous layer. A schematic of the bilayer sample is shown in the inset.
This project funding has provided the opportunity for the principal investigator(PI) and his students to initiate studies with XPCS, a technique hardly known in the polymer community when the original proposal was written. This has opened up other opportunities for the study of polymer surface dynamics for the PI and his research group which he intends pursue with requests to other funding agencies. The collaboration with a theorist which developed in the course of the project has also greatly enriched the research experience of the graduate student.
References:
1) Zhao, B.; Brittain, W. J.; Zhou, W.; Cheng, S. Z. D. J. Am. Chem. Soc. 2000, 122, 2407



