Reports: G4
46807-G4 Controlled Electron Transfer in Hydrogen Bonded Systems
The aim of our research is to improve the effectiveness of photoinduced charge-separation between donors and acceptors. The long-lived charge separated states will then have enough time to undergo desired chemistry. At the beginning of this project, we investigated hydrogen-bonded naphthalimide-pyridine (NI-PYR) systems, with a goal to learn how to take advantage of H-bond dynamics to control the electron flow in molecular systems. To obtain a good understanding of the chromophore used in our donor-acceptor system, we started out with the investigation of excited state properties of five NI derivatives.1 For this purpose, my group developed a femtosecond pump-probe instrument that can probe the transient species in both the visible and mid-IR range. 1-6 One of the main findings of our work is that the excited state properties of NI derivatives are characterized by a competition between n,p* and p,p* excited states. We further studied electron transfer dynamics from excited SMe-NI to NO2,CN-PYR.2 The main finding of our work is that the fast deactivation of the excited state occurs in the H-bonded complex. Unfortunately, the fast thermal deactivation of the SMe-NI excited state in H-bonded complex made it impossible to study electron transfer dynamics in NI-PYR systems.
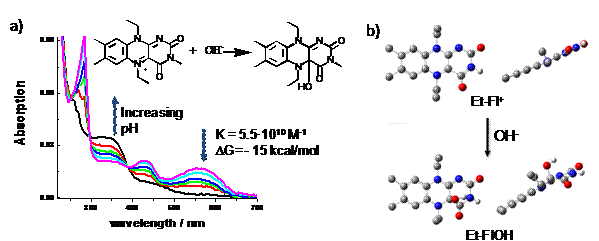
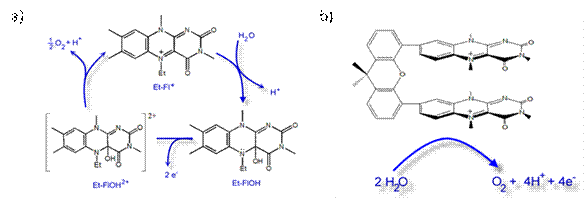
This initial study led us to develop a project aimed at using flavonium salt derivative for catalytic oxidation of water. The main goal of this research is to use proton-coupled electron transfer to drive the production of peroxyl radicals from a flavin-based alcohol. The further oxidation of peroxyl radicals is expected to produce oxygen and recover the flavonuim salt.
The reaction of Et-Fl+ with
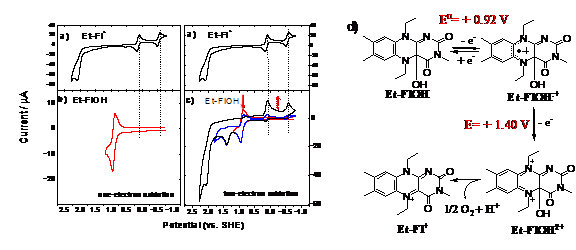
To investigate the behavior of Et-FlOH upon one- and two-electron
oxidation, we performed cyclic voltammetry using acetonitrile as a solvent. The
cyclic voltammogram of Et-Fl+
consists of two reversible one-electron reductions at E1/2= + 0.13 V
and E1/2 = -0.54 V. Furthermore, Et-Fl+ shows an irreversible one-electron oxidation at
2.27 V. Figures 2b and c show cyclic voltammograms of Et-FlOH. The scan up to + 1.2 V reveals a reversible one-electron
oxidation peak at E1/2=+0.92 V (red curves), suggesting that the
radical cation of Et-FlOH is stable over the course of the cyclic voltammetry
scan (scan rate: 100 mV/s). As we scan to higher potentials (blue and black
curves), we observe an irreversible oxidation peak at + 1.4 V, that corresponds
to the two-electron oxidized Et-FlOH2+.
Upon this irreversible two-electron oxidation, we observe a decrease in the
current originating at +0.92 V and an increase of new peaks at +2.27 V, +0.13
and -0.54 V, all of which are signature peaks of Et-Fl+. These peaks are consistent with the conversion
of Et-FlOH2+ to Et-Fl+. Figure 2d outlines
the processes occurring at the electrode. The conversion of Et-FlOH2+ to Et-Fl+ suggests that the oxygen
is most probably released in the process.
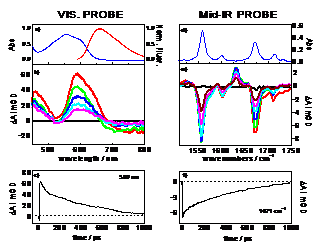
We are currently investigating the photophysical
behavior of Et-FL+ and Et-FlOH using pump-probe spectroscopy. Figure below
presents visible and mid-IR transient absorption spectra of Et-Fl+. The visible spectrum
consists of excited-state absorption (600 nm), ground-state bleach (520 nm) and
stimulated emission (750 nm) signals that arise from 1S state of Et-Fl+. The TRIR spectra of Et-Fl+ show signals due to
C=N and C=O vibrations of Et-Fl+ in
its excited state. The future set of experiments
will involve photochemical oxidation of Et-FlOH in the presence of a sacrificial
acceptor.
Reference:
(1) P. Kucheryavy, G. Li, S. Vyas, C. Hadad, K. D. Glusac, " Electronic Properties of 4-Substituted
Naphthalimides", J. Phys. Chem A, 2009, 113, 6453-6461.
(2) P. Kucheryavy, G. Li, S. Vyas, C. Hadad, K. D. Glusac,
"Ultrafast Excited State Deactivation in Hydrogen-Bonded
Naphthalimide-Pyridine Donor-Acceptor Systems", In Preparation.
(3) G. Li, K. Parimal, S. Vyas, C. M. Hadad, A.
H. Flood, K. D. Glusac, Pinpointing
the Extent of Electronic Delocalization in the Re(I)-to-Tetrazine Charge
Separated Excited State Using Time-Resolved Infrared Spectroscopy, J. Am. Chem. Soc., 2009,
131, 11656-11657.
(4) G. Li, K. D. Glusac, The Role of
Adenine in Fast Excited State Deactivation of FAD: A Femtosecond mid-IR
Transient Absorption Study, J. Phys. Chem. B., 2009, 113,
9059-9061.
(5) G. Li, V. Sichula, K. D. Glusac, The Role of Adenine in Thymine Dimer Repair by
Reduced FAD, J. Phys. Chem. B, 2008, 112, 10758-10764.
(6) G. Li, K. D. Glusac Light-Triggered Proton
and Electron Transfer in Flavin Cofactors, J. Phys. Chem. A, 2008,
112(20), 4573-4583.