Reports: G3
48410-G3 High Oxidation State, Late Transition Metals Featuring Metal-Ligand Multiple Bonds: Strategies for Sequential C-H Bond Activation and Functionalization
Summary. In the field of homogenous catalysis, few reports have appeared offering general, chemically mild methods for the introduction of nitrogen- or oxygen-containing functional groups into simple hydrocarbon substrates. The combination of small-molecule activation with CH bond functionalization represents a significant advance in converting inexpensive chemical feed stocks (e.g. hydrocarbons) to valuable functional molecules with minimal or complete absence of waste generation. We are developing multi-electron redox transformations by mid-to-late, first row transition metal complexes targeting methods for the functionalization of CH bonds. We have synthesized a new class of electrophilic complexes featuring transiently-formed, or metastable metal-ligand multiple bonds capable of mediating CH functionalization. The hard donor, weak-field pyrrole-based ligand scaffolds were selected for this study primarily for their ability to confer a range of steric and electronic environments to the bound transition metal ion. The coordination chemistry was investigated for the redox-active dipyrromethane and tris(pyrrolyl)ethane supported transition metal complexes to establish the electronic structure for the pyrrole-based ligand donors. Dipyrromethene (or semi-porphyrin) supported ferrous complexes were found to effect a range of intra- and intermolecular CH bond functionalization reactions - allowing for the construction of new CN, CC and CO bonds from unactivated CH bonds, culminating in catalytic amination reactions. While electron-releasing ligands (phosphines, carbenes, and β-diketiminates) have been elegantly employed to stabilize metal-ligand multiple bond formation on mid-late transition metal platforms, we posit that the weak-field ligand environment is critical to the overall efficacy of the dipyrromethene-supported catalyst system. The following is a brief summary of the research performed during the PRF funding period during the past 24 months.
 Coordination
chemistry of pyrrole-based ligand scaffolds (Specific Aim #1).
Coordination
chemistry of pyrrole-based ligand scaffolds (Specific Aim #1).
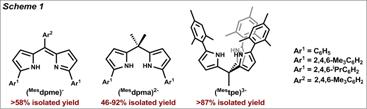
Our strategy to effect sequential bond activation and functionalization requires a ligand system or systems that (1) confer stability to metal ions over a range of possible oxidation states (e.g. MII ↔ MIV), (2) maintain a minimal coordination environment at the metal center and create a sterically enforced pocket to protect metal-ligand multiply-bonded linkages, and (3) create an electrophilic metal environment (limit ligand electron-donicity). Pyrrole-derived ligands (Scheme 1): monoanionic dipyrromethene, dianionic dipyrromethane and trianionic tris(pyrrolyl)ethanes are good candidates to satisfy all of the desired design criteria. The pyrrole substituents exhibit attenuated π-basicity of the nitrogen donors from conjugation with the pyrrole π-system, satisfying criterion 3 and making these platforms more amenable for coordination to late transition metal ions.
Figure 1 shows representative transition metal complexes of the three primary ligand platforms used in this study. In this fashion, divalent complexes of Mn→Zn featuring the monoanionic dipyrromethene, dianionic dipyrromethane, and trianionic tri(pyrrolyl)ethane ligands have been prepared. In general, the pyrrole groups are found to coordinate in an η1-fashion, accommodating a range of coordination geometries at the metal ion as confirmed by X-ray diffraction studies. Given the weak-field nature of the pyrrolide donors, the resulting paramagnetic complexes were characterized by standard spectoscopic means (1H NMR, UV-Vis, SQUID magnetometry, EPR where appropriate, Mössbauer for Fe-containing complexes, cyclic voltammetry, mass spectrometry, and combustion analysis).
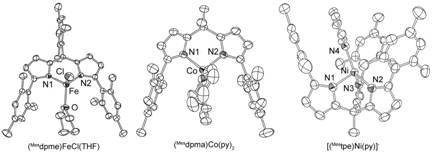
Figure 1. Solid state molecular structures for (left) (Mesdpme)FeCl(thf), (center) (Mesdpma)Co(py)2, and (right) [MestpeNi(py)] (thermal ellipsoids set at 50%).
The dianionic (Mesdpma) ligand afforded high-spin, pseudo-tetrahedral complexes for MnII, FeII, and CoII, but afforded a low-spin, square-planar NiII with pyridine as the ancillary ligands. Despite the ligand's dianionic nature, the pyrrolide donors proved to be weak σ-donors in addition to being poor π-donors. Surveying the (Mesdpma)M(py)2 complexes by differential pulse voltammetry revealed two one-electron oxidation events (Figure 2a), indicating the oxidations are independent of the bound metal, its spin state or geometry (i.e., the voltammagrams neatly overlay for the CoII, NiII and ZnII complexes). Thus we concluded the origins of the redox for the (Mesdpma) complexes is entirely ligand based (corroborated by DFT analysis), indicating there is very little covalency between the (Mesdpma) platform and the bound metal ion. The dipyrromethene (Mesdpme) is a very intense chromophore, akin to their porphyrin analogues. Binding a transition metal ion causes a very modest red-shift in the ligand π→π* electronic transition (Figure 2b). Cyclic voltammetry on (Mesdpme)FeCl(THF) reveals a fully reversible FeIII/II redox wave centered at -400 mV (Figure 2b).
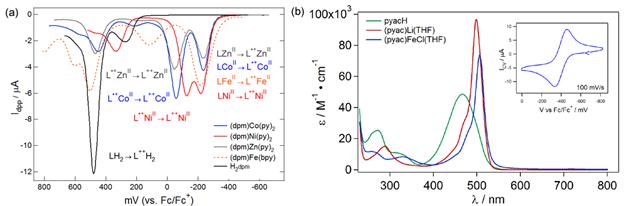
Figure 2. (a) Differential pulse voltammagrams of solutions in THF of (Mesdpma)M(py)2; (b) UV-vis spectra of (Mesdpme)M for M = H, Li(THF), FeCl(THF); inset displaying CV for (Mesdpme)FeCl(THF).
Reactivity Studies. The dipyrrolide redox activity in (Mesdpma)M complexes has precluded our attempts to observe oxidative group transfer reactions. However, the more redox-stable dipyrromethene complexes have proven to be well-suited to canvassing reactivity. We have shown the (Mesdpme)FeCl(THF) complex to be active for a range of intramolecular group transfer reactions. For example, the complex readily decomposes organic azides (electron-rich and electron-poor) to afford products consistent with formal nitrene delivery to a ligand CH bond (Scheme 2). The stoichiometric reactions are easily followed by 1H NMR, as the desymmetrization of the ligand due to the CH bond amination is readily apparent even with the paramagnetically shifted proton resonances. Reaction of (Mesdpme)FeCl(THF) with dioxygen does afford a small amount of ligand hydroxylation, but the reaction was substantially improved (nearly quantitative) when the urea-H2O2 was used as an O-atom source. Reaction of (Mesdpme)Fe(OTf)(THF) with several equivalents of N2CHCO2Et showed that 1-4 carbene additions (:CHCO2Et) to the ligand framework occurred by ESI-MS, with the majority of the products showing 3 carbene insertions.
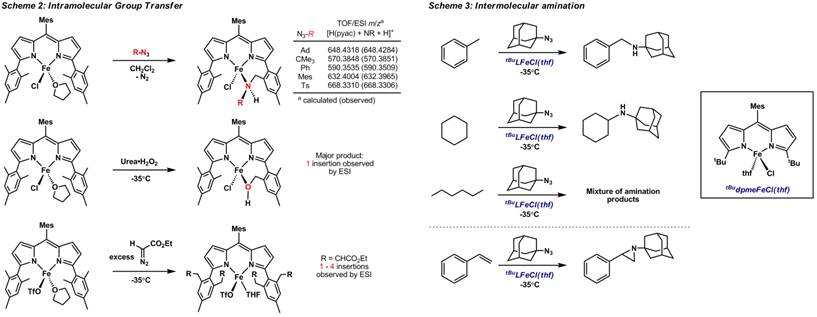
Intermolecular CH bond amination. To generate a dipyrromethene Fe-complex capable of intermolecular CH activation and functionalization, we substituted the (Mesdpme) mesityl flanking units with tert-butyl substituents. Reacting (tbudpme)FeCl(THF) with one equivalent of 1-azidoadamantane at -35°C in toluene (BDE: 89.7 kcal/mol), cyclohexane or linear hexane (BDE: 95.5 kcal/mol), or styrene consumes the azide and cleanly affords the respective secondary amine or aziridine (from styrene) products (Scheme 3, products analyzed by 1H NMR, GC-MS). Preliminary reactivity screens suggest the intermolecular CH bond functionalization could be carried out under the non-optimized catalytic conditions using alkyl azides (tBuN3, AdN3) with 2.5 mol% (tbudpme)FeCl(THF) in neat hydrocarbons at 50-60°C.



