Reports: AC3
46121-AC3 Systematic and Theoretical Studies of Anion-pi Interactions for the Development of Supermolecules and New Materials
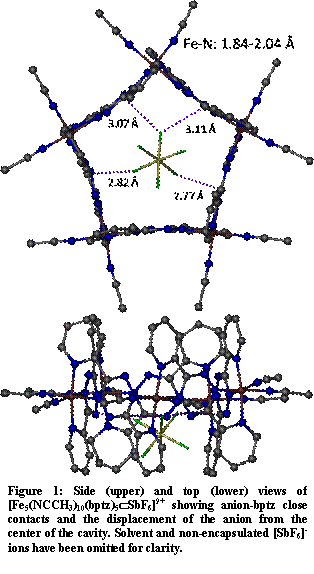 Figure 1: Side (upper) and top (lower) views of [Fe5(NCCH3)10(bptz)5ÌSbF6]9+ showing anion-bptz close contacts and the displacement of the anion from the center of the cavity. Solvent and non-encapsulated [SbF6]- ions have been omitted for clarity.
Figure 1: Side (upper) and top (lower) views of [Fe5(NCCH3)10(bptz)5ÌSbF6]9+ showing anion-bptz close contacts and the displacement of the anion from the center of the cavity. Solvent and non-encapsulated [SbF6]- ions have been omitted for clarity.Work has continued on extending the family of anion-templated square and pentagonal architectures to other firstrow transition metals, most notably iron. We have recently synthesized both the square and pentagonal architectures with Fe(II) following the same preparatory procedure as in the Ni(II) square and pentagonal motifs. X-ray diffraction analysis shows that the cavity of both the square and pentagonal motif are contracted when compared to the Ni(II) analog. Anion-pi interactions between the anion and the central ring of 3,6-bis(2'-pyridyl)-1,2,4,5-tetrazine (bptz) ligand are present in both cases. In the Fe(II) square motif, the [BF4]- anion is not centrally located in the cavity due to the contracted cavity size, yet close contacts between the anion and the carbon atoms of bptz are still evident, and the anion remains within the confines of the cavity. In the Fe(II) pentagonal motif, the [SbF6]- anion maximizes close contacts between the anion and the electron-deficient carbon atoms of bptz, with the closest contact distances being between the carbon atoms of the tetrazine core and the fluorine atoms of the hexafluoroantimonate.
Bond distances from between Fe(II) and the nitrogen atoms surrounding it, as well as preliminary magnetic studies, suggest that the iron(II) ions in both motifs are primarily in the low-spin state. NMR studies have begun with both the pentagonal and square motifs, and preliminary results show resonance shifts different than those of free bptz, as expected. Also, the resonances are sharp, lending further support to the low-spin assignment of Fe(II).
Recently, a target of interest in our laboratories has been the HAT(CN)6 molecule (1,4,5,8,9,12-hexaazatriphenylene-hexacarbonitrile; Figure 1A) which is an extended p electron-deficient molecular unit with multiple sites available for potential interactions with anions (electron deficient areas of the molecule are indicated by the blue areas in the electrostatic potential map; Figure 1B).
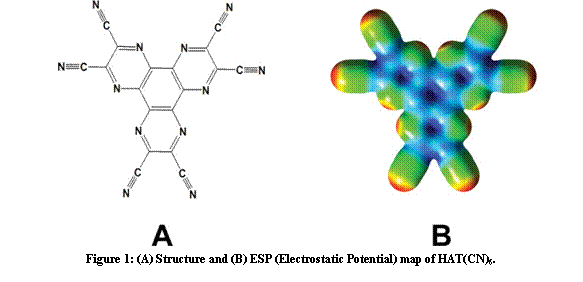 Figure 2: (A) Structure and (B) ESP (Electrostatic Potential) map of HAT(CN)6.
Figure 2: (A) Structure and (B) ESP (Electrostatic Potential) map of HAT(CN)6.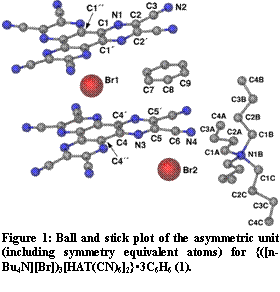 Figure 3: Ball and stick plot of the asymmetric unit (including symmetry equivalent atoms) for {([n-Bu4N][Br])3[HAT(CN)6]2}3C6H6 (1).
Figure 3: Ball and stick plot of the asymmetric unit (including symmetry equivalent atoms) for {([n-Bu4N][Br])3[HAT(CN)6]2}3C6H6 (1).Previous work performed in our group demonstrated that HAT(CN)6
is capable of co-crystallizing in the presence of [n-Bu4N][I] to form the intensely colored dark
green/black crystals of
Quantitative analyses of the intensities of the new
absorption bands at 630, 419 and 408 nm for the iodide,
bromide and chloride complexes in THF, respectively, as a function of the
respective halide [X-] concentration were performed,
given the 2:3 stoichiometric ratio of [HAT(CN)6]:[halide] in solution from the Job plots that were performed.
The determined association constant values increase
in the order KCT,Cl >
KCT,Br > KCT,I. The
spontaneous formation of
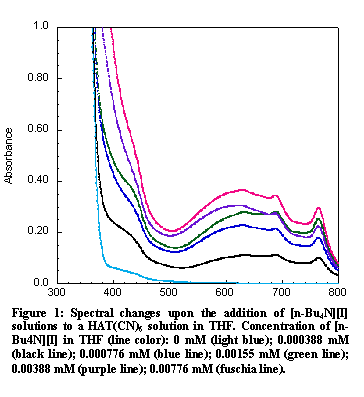 Figure 4: Spectral changes upon the addition of [n-Bu4N][I] solutions to a HAT(CN)6 solution in THF. Concentration of [n-Bu4N][I] in THF (line color): 0 mM (light blue); 0.000388 mM (black line); 0.000776 mM (blue line); 0.00155 mM (green line); 0.00388 mM (purple line); 0.00776 mM (fuschia line).
Figure 4: Spectral changes upon the addition of [n-Bu4N][I] solutions to a HAT(CN)6 solution in THF. Concentration of [n-Bu4N][I] in THF (line color): 0 mM (light blue); 0.000388 mM (black line); 0.000776 mM (blue line); 0.00155 mM (green line); 0.00388 mM (purple line); 0.00776 mM (fuschia line).Computational studies were undertaken in order to better understand the halide···HAT(CN)6 interactions. Inspection of the crystal structures of either the Br or I complex reveal two distinct crystallographic positions occupied by the halide anions. The first is situated above the centroid of the HAT(CN)6 molecule, henceforth referred to as the central position, and the second is situated above the peripheral pyrazinyl C-C bond of the molecule, or the outer position (see Figure 3). Single anion single HAT(CN)6 geometry optimizations where performed using Density Functional Theory (DFT) with Cl- and Br- anions. The resulting structures were then analyzed using Natural Bond Orbitals (NBO) (Table X1). The NBO analysis reveals that there is a greater degree of CT present between the HAT(CN)6 molecule and the Cl- anion then with the Br- anion. The analysis also revealed that there is a greater degree of CT for the outer position then the central position. These results are consistent with spectroscopic data presented above.
| Stabilization energy arising from CT interactions as predicted by NBO. | |
|---|---|
|
HAT(CN)6 with
|
ΔE (kcal/mol)
|
|
Cl (central)
|
13.54
|
|
Cl (outer)
|
25.66
|
|
Br (central)
|
6.6
|
|
Br (outer)
|
19.97
|



