Reports: G1
47698-G1 Binuclear Metal Complexes as Asymmetric Catalysts
The activation of relatively unreactive C-H bonds for C-C or C-X bond formation frequently relies on the intervention of organometallic intermediates and often involves oxidative processes. In the course of pursuing our originally proposed research, we have discovered a new alpha-functionalization reaction of cyclic amines that proceeds without the involvement of transition metals or other additives (Figure 1). In this redox-neutral condensation reaction, a comparatively unreactive C-H bond alpha to a ring-nitrogen is replaced by a C-N bond, concomitant with the formation of a new ring system. This thermally promoted reaction between an aminoaldehyde and a cyclic amine results in the formation of a ring fused aminal and thus provides convenient access to this structural motif. The reaction conditions resemble those used in Friedlaender condensations of aminoaldehydes and ketones to form quinolines in which pyrrolidine is frequently used as a base promoter. Some of these aminals represent reduced versions of quinazolinone alkaloids, compounds that have attracted significant attention in the synthetic community due to their diverse array of biological activities. Selective oxidation of ring fused aminals provides rapid access to this structural motif which allowed for the synthesis of the natural prodcuts deoxyvasicinone and rutaecarpine in just one additional step.
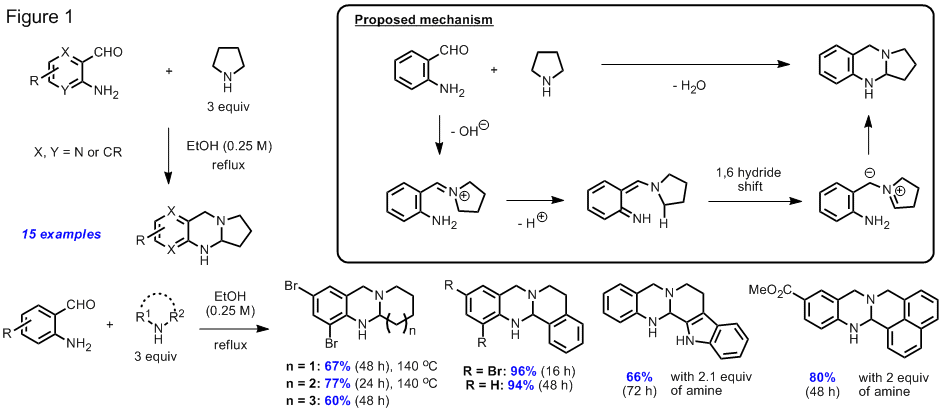
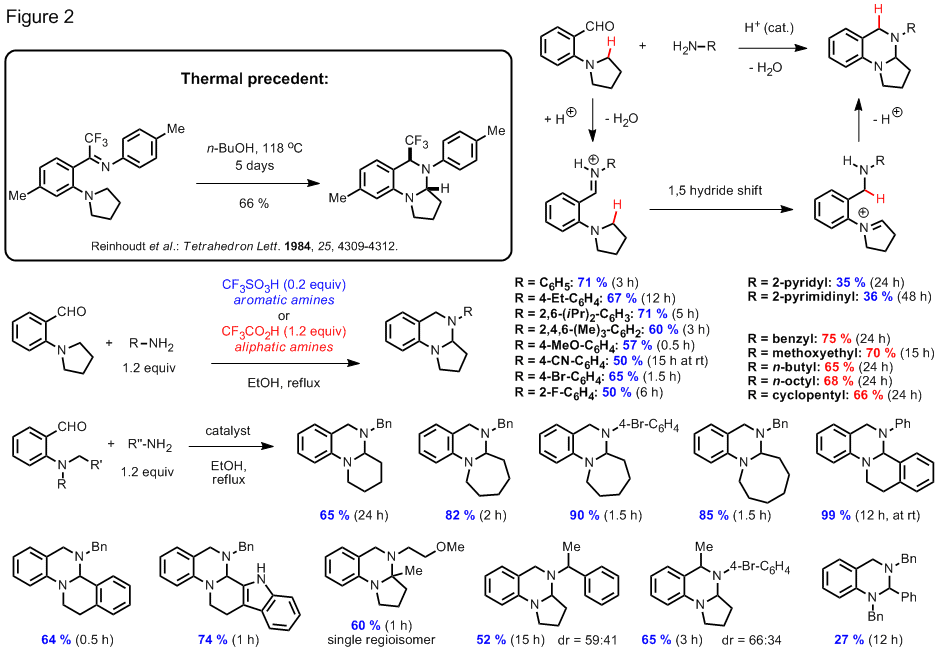
We have succeeded in preparing other aminal products that represent regioisomers of the compounds shown in Figure 1. In an overall redox neutral process that is believed to involve a 1,5-hydride shift event, aminobenzaldehydes are condensed with aliphatic or aromatic amines in an acid promoted process. The scope of this reaction is summarized in Figure 2. Reactions of aminobenzaldehydes with aromatic amines were found to proceed well in the presence of catalytic amounts of triflic acid whereas the corresponding reactions with aliphatic amines worked best when an excess of trifluoroacetic acid was employed.
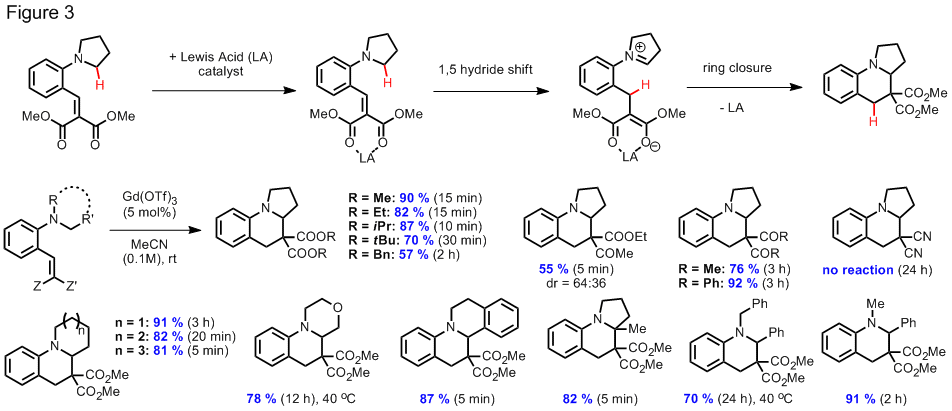
In a related study, we have found that catalytic amounts of various Lewis acids facilitate rearrangement of alkylidene malonates to complex tetrahydroquinolines at room temperature (Figure 3). Scandium triflate readily catalyzed this transformation. Albeit less efficiently, several main group and transition metals were also found to catalyze this rearrangement. Remarkably, gadolinium triflate showed striking rate acceleration as compared to scandium triflate (up to 88 fold rate acceleration under optimized conditions). Again, a 1,5 hydride shift is thought to be involved in this redox neutral reaction. Tetrahydroquinolines are at the core of many biologically active materials and the compounds prepared in our study would be difficult to obtain by other means. This work was highlighted in the March 2009 issue of Synfacts.
This PRF grant has supported a number of graduate students over the past year. In addition, funding by the PRF enabled us to obtain a sufficient amount of preliminary data on this project, ultimately resulting in funding from the NSF. The originally proposed research is still subject of ongoing investigations and is expected to result in at least two future publications that will acknowledge PRF support.



