Reports: AC3
48286-AC3 Characterization of Actinide - Ligand Bonding
Chemical bonding may be described in terms of the distribution of the valence electrons in a molecule. The X-ray diffraction experiment, through the Fourier transform of measured structure factors, contains the distribution of the total electron density modified by thermal motion. For light atom structures, the valence electron count is a significant part of the total electron count. However, for elements as heavy as the actinides, the X-ray scattering is dominated by the core electrons. Hence, even for quite simple compounds, the requirements on data quality are extreme. In addition, although the modeling of the electron density via a multipolar expansion for light atoms is well established, there is no body of knowledge regarding the best functions to use for heavy elements. In this funding period, we have prepared and crystallized a number of simple actinide compounds, made significant improvements to the data integration software, improved our program for absorption corrections, and tested a number of approaches to the multipole refinements.
Crystals have been obtained for the following simple compounds Cs2ThCl6, Cs2UCl6, (Et4N)2ThCl6, NaUO2(OAc)3, (Et4N)4Th(SCN)8, Th(S2PMe2)4. Crystal quality is critical to these extreme experiments, hence their quality is currently being evaluated prior to any low temperature studies.
We had previously reported a proof of principle experiment on Cs2UO2Cl4 crystals. A new data set has now been collected on a better crystal at 20 K. This repeat measurement was carried out as the previous data, despite giving an encouraging refinement, still had a rather noisy residual map. The refinement using this data is currently incomplete.
Following concerns that our previous data integration, despite giving excellent data for light atom structures, was still not adequate for studies on heavy atoms, a new approach to profile fitting of the diffraction data has been implemented into the VIIPP integration program. Benchmarking with light atom data sets indicates that there is significant improvement, particularly in the weak, high angle data. This is now being tested on the Cs2UO2Cl4 data sets.
For heavy atom structures, the correction of the observed data for the effects of absorption is essential, even for the very small crystals that we are using (~60μ). We have hence made some improvements both to our absorption program, and to the hardware for measuring the crystal size and shape.
Within the XD program that we use for fitting the multipole electron density model, there are many options for building the model for an atom as heavy as uranium. None of these has been tested with real data, hence we have spent a significant amount of time exploring different refinement strategies. At the same time, we have discovered several errors in this untested part of the program, and have been communicating with the programmers to rectify these bugs. Until these corrections have been made, we do not have a best strategy.
The deformation and Laplacian maps shown below obtained from the current stage of refinement provide an indication of the progress made. Although this is still a work in progress, we are very encouraged by this result.
I note that Dr. Deepak Chopra who worked on this project has accepted a faculty position at the Indian Institute of Technology in Bhopal, India.
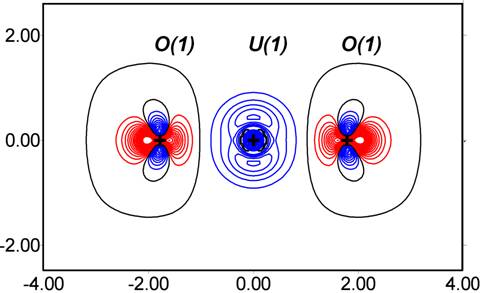
UO22+ deformation density
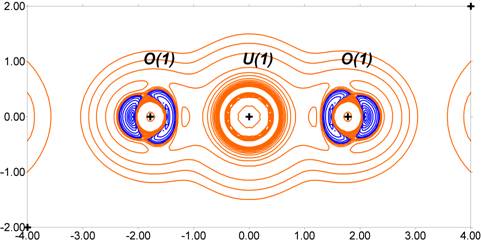
UO22+ Laplacian of the total electron density



