Reports: AC10
47379-AC10 First-Principles Studies of Water Splitting at Semiconductor and Oxide Surfaces
Introduction
The need to decrease the dependence on energy from carbon-based sources is widely recognized and renewable-energy alternatives are actively being pursued. Among these the most promising technologies are wind turbines and solar cells. However, these sources deliver electricity literally only as the wind blows or the sun shines. Thus, efficient energy storage will be essential in a society where a significant fraction of energy is delivered by renewable sources. One possibility is to convert electrical into chemical energy using electrolysis, splitting water into hydrogen and oxygen.
In the present project we go one step further and study direct hydrogen production by a photoelectrochemical cell, which combines a solar cell and an electrolysis cell into one device and produces hydrogen directly without the loss inherent in the traditional two-step process. One of the electrodes in a photoelectrochemical cell consists of a semiconductor that absorbs sunlight and produces electrons and holes, which split water into hydrogen and oxygen. In order to be efficient, the semiconductor must fulfill at least three criteria: 1) the material must be stable under illumination, 2) the band gap must be such that a significant fraction of the solar spectrum is absorbed, and 3) the valence- and conduction-band edges must straddle the redox potential for both hydrogen and oxygen evolution.
We choose to investigate alloys of GaN and InN since these are known to be stable against corrosion, and thus likely to fulfill criterion number 1). Band gap of InGaN
In order to address criterion 2), i.e., to choose an alloy composition that leads to absorption of a sizeable fraction of solar photons, one must know the band gap as a function of In content. Considerable research efforts have already been devoted to determining the band gap as a function of In content. However, experimental measurements suffer from the decline in materials quality at higher In compositions, and are complicated by the presence of strain in thin films. A number of theoretical studies have also been reported, all based on density functional theory (DFT). However, DFT suffers from the well-known band-gap problem and therefore conventional DFT results have large uncertainties. In the present study we take advantage of a recent breakthrough in exchange-correlation (XC) functionals, using a hybrid functional (in particular the HSE06 functional developed by Heyd, Scuseria, and Enzernhof) that provides considerably more accurate band gaps than previous XC-functionals, while maintaining excellent accuracy in energetic and structural results.
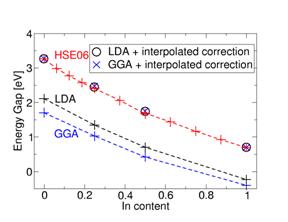
Figure 1 Band gap as a function of In content, calculated using different exchange-correlation functionals. Also shown are the results of correction schemes wherein the GaN and InN band gaps are adjusted and the corrections for intermediate compositions are linearly interpolated.
The band gap of InGaN alloys obtained with different XC functionals is shown in Figure 1. Band gaps of alloys are often fitted with an equation of the form Egap(x) = (1 - x)Egap(GaN) +xEgap(InN) - bx(1 - x), where b is the so-called bowing parameter. We find that the best overall fit is obtained with b=1.10 eV; however, at low In contents the bowing is much stronger and at high In content it is weaker. Therefore, it is necessary to use an In-content-dependent bowing parameter. Finally we find that alloy band gaps can be interpolated with satisfactory accuracy (less than 0.05 eV deviation from the full results) based on less computer-intensive XC functionals such as the local density approximation (LDA) or the generalized gradient approximation, along with corrected (HSE06) band gaps for the pure nitrides.
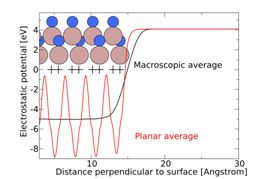
Figure 2 Electrostatic potentials obtained from surface calculations for the m plane of GaN. Planar averaged potentials as well as macroscopically averaged potentials are plotted in the direction perpendicular to the surface. Plus signs mark the position of atomic planes. Position of band edges
Criterion 3) requires that the valence- and conduction-band edges straddle the redox potential of oxygen and hydrogen evolution. Thus, the band alignments on an absolute energy scale (e.g., with respect to the vacuum level) must be known. We obtain this alignment from surface calculations, which link the electrostatic potential inside the bulk solid to the vacuum potential, as seen in Figure 2. Calculations of the bulk band structure then provide information about the position of valence and conduction bands with respect to the electrostatic potential.
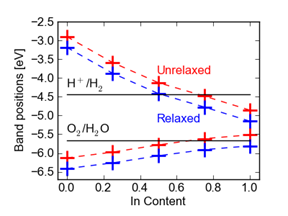
Figure 3 InGaN band alignments relative to vacuum.
The resulting band alignments relative to vacuum for InGaN are shown in Figure 3. The dependence of valence-band position on In content is very close to linear; all the nonlinearities (bowing) in the band gap are contained in the conduction band. Surface composition
In addition to our investigations of the electronic structure of InGaN alloys we are also investigating the surface composition as a function of reaction conditions and valence-band positions. We find that a surface oxide layer exists on the n-type nitrides and that these surface oxide layers result in modest overpotentials for water splitting. A detailed description of the surface composition will form the basis for investigating the chemistry of water splitting or hydrogen evolution at InGaN alloy surfaces. Summary
We have demonstrated that DFT calculations based on advanced functionals (such as the HSE06 hybrid functional) produce band gaps in close agreement with experiment, which is essential for accurately calculating alloy band gaps and band alignments on an absolute energy scale. Our results thus indicate that DFT with HSE06 can give valuable insight into key properties in photoelectrochemistry. Finally, we find that GaN has modest overpotentials for water splitting. Future work will include further investigations of surface reconstructions, including oxide layers, and the effect on hydrogen evolution.



