Reports: G1
46157-G1 Chiral Acyclic Carbene Ligands for the Asymmetric Suzuki Coupling Reaction
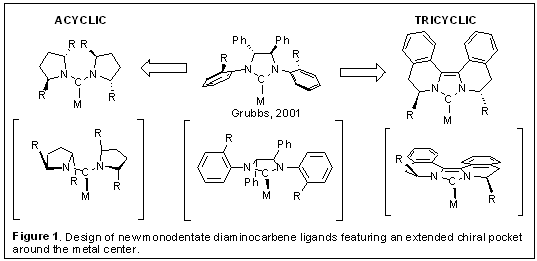
The goal of this research is to develop novel chiral monodentate diaminocarbene ligands for asymmetric catalysis. We have designed novel acyclic diaminocarbenes featuring C2-symmetric pyrrolidines as well as the tricyclic biisoquinoline-based carbenes with an idea of creating a chiral environment close to the metal center by positioning stereogenic centers at the positions α to the nitrogens (Figure 1). The asymmetric Suzuki coupling reaction was initially selected to evaluate new chiral ligands because we envisioned that the highly electron donating and sterically demanding nature of acyclic carbenes would be beneficial for the Suzuki coupling reaction.
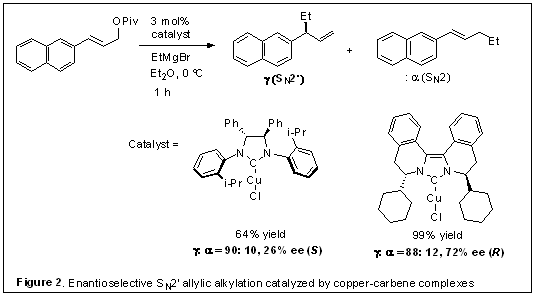
1. Biisoquinoline-based tricyclic chiral diaminocarbene ligands. In our last annual report, we described the development of biisoquinoline-based tricyclic chiral diaminocarbene ligands (BIQ) and their Pd and Cu complexes. The chiral environment is created in close proximity to the metal center, which is confirmed by an X-ray crystal structure. The concise ligand synthesis is highlighted by a modified Bischler-Napieralski cyclization of bisamides prepared from readily available chiral phenethylamines, and allows easy variation of the stereodifferentiating groups. It was found that the cyclohexyl-BIQ-copper complex is an efficient catalyst for enantioselective SN2' allylic alkylation with Grignard reagents, showing SN2' regioselectivity higher than 5:1 and enantioselectivity in the range of 68-77% ee (Figure 2).
In an effort to further improve enantioselectivity, we developed new C1-symmetric isoquinoline-based ligands over the past year. This novel C1-symmetric bicyclic ligand is designed to block three quadrants of metal coordination sphere, which was confirmed by an X-ray crystal structure of the metal complex. This C1-symmetric isoquinoline-based ligand again features a stereogenic center at the position α to the nitrogen atom. Unfortunately, the copper (I) complex with the new C1-symmetric ligand showed lower enantioselectivity (35% ee) in allylic alkylation with Grignard reagents. However, recently we discovered that high enantioselectivity (>82% ee) was observed in Cu-catalyzed β-boration reactions where C2-symmetric BIQ-Cu catalyst showed much lower ee, and this result will be published in due course. This example demonstrates importance of access to a diverse pool of chiral ligand candidates. The two new chiral NHC ligands that we have developed lead to improved stereoselectivity in different transformations, and they provide more options in terms of selecting monodentate chiral NHCs for new asymmetric transformations.
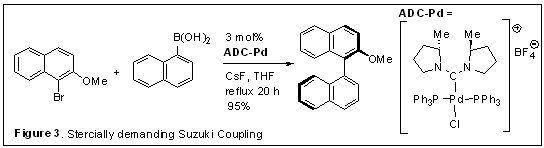
2. Acyclic diaminocarbene ligands. Our initial efforts were directed towards preparation of chiral acyclic diaminocarbenes featuring 2-substituted pyrrolidines as a model system. Chiral ADC Pd complexes were conveniently prepared through oxidative addition of chloroamidinium ions by Pd(PPh3)4, and the ADC-Pd complexes were active catalysts for sterically demanding asymmetric Suzuki coupling reactions (Figure 3). However, observed enantiomeric excesses were low (5-7% ee) presumably owing to the conformational flexibility, suggesting that incorporation of C2-symmetric 2,5-trans-dialkylpyrrolidine units might be critical in order to obtain high enantioselectivity.
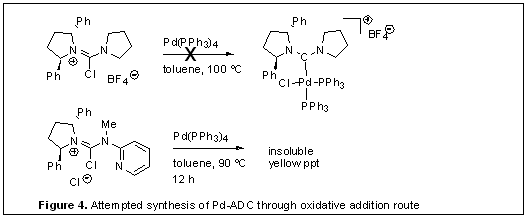
Our attempts at synthesis of a N,N,N′,N′-tetra-alkyl-substituted urea and corresponding chloroamidiniums were unsuccessful, and a stable carbamoyl chloride compound was isolated after the reaction of 2,5-trans-diphenylpyrrolidine with phosgene. In addition, preparation of chiral ADC-Pd complexes using oxidative addition protocol from even less sterically demanding acyclic carbenes, for example, an ADC stemming from one 2,5-trans-diphenylpyrroldine and a nonsubstituted pyrrolidine were not successful (Figure 4). This result illustrates a drawback of oxidative addition protocol where phosphine ligands are necessary for the key step. This limitation motivated us to develop an alternative route to various ADC-metal complexes especially with relatively bulky chiral ADC ligands. Recently we identified a more efficient synthetic route to various chiral acyclic carbenes that overcame the aforementioned problems. Several chiral ADC-metal complexes have been prepared and characterized by X-ray. Testing those new chiral ADC-metal complexes in asymmetric catalysis is currently in progress.
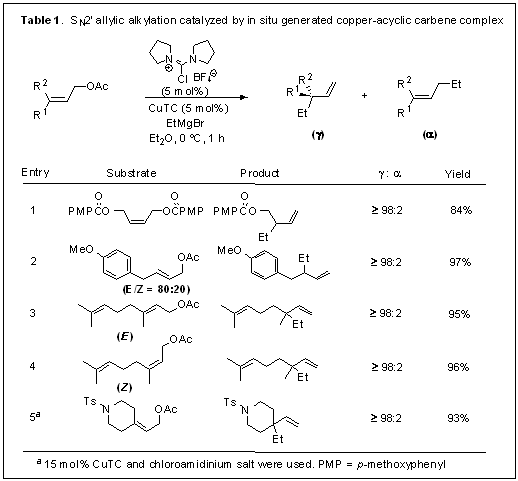
Over the past year, we have discovered a new route to ADC-Cu complexes using chloroamidiniums as a convenient carbene precursor (published in Chem. Commun.). A novel acyclic diaminocarbene-copper complex is generated from a chloroamidinium salt and Cu(I)-thiophenecarboxylate in the presence of Grignard reagent. 13C NMR experiments using 13C-labeled carbene precursors suggest the generation of an acyclic diaminocarbene-copper species. This in situ generated acyclic diaminocarbene-Cu complex is a highly efficient catalyst for SN2′-allylic alkylation with alkyl Grignard reagents, showing excellent γ selectivity for various allylic substrates. Symmetrical dibenzoate substrates can be used owing to high γ selectivity, and quaternary centers can be generated from tri-substituted alkene substrates in high yield (Table 1). The reaction with nitrogen atom containing substrates was sluggish and 15 mol% of catalyst loading was required. However, this ADC-Cu catalyst appears to be more reactive than the NHC-Cu catalyst (IMesCuCl) which gave 24% yield under identical conditions. Future work will focus on development of enantioselective allylic alkylation using chiral ADC-Cu complex.
The ACS PRF grant has been invaluable to initiate our research program. Thanks to the early support by ACS-PRF, we received a three year research grant from the Florida Department of Health, James & Esther King Biomedical Research Program.



