Reports: AC7
47700-AC7 ROMP Healing Agent Development for Self-Healing Materials
In the past decade, polymers and composites that can repair themselves with autonomy have been extensively studied in academia and received significant commercial interest [[1]]. Perhaps the most successful self-healing mechanism developed to date incorporates liquid healing agent-filled microcapsules and catalyst particles into a polymer matrix. Upon material fracture, microcapsules rupture, followed by flow of the liquid healing agent into the crack volume. When the healing agent contacts the catalyst particles it polymerizes and adheres the crack faces together [[2]]. The most well-studied, and probably most successful, healing agent/initiator combination used thus far in self-healing systems is Dicyclopentadiene (DCPD) and Grubbs' catalyst, the former of which undergoes a reaction called ring-opening metathesis polymerization (ROMP) in the presence of the latter. This healing agent/catalyst system has been shown to only partially self-repair a fractured composite, and many factors have been identified as responsible for this weak healing ability (discussed in more detail below). However, very little work has been done to modify and optimize the DCPD/Grubbs' catalyst system to improve the self-healing capability. Hence, in this work we systematically develop novel healing agent/catalyst combinations that satisfy the many unique requirements of a self-healing system. In addition, many of our experimental results can be extrapolated to develop models useful for predicting the self-healing performance of a wide range of healing agents.
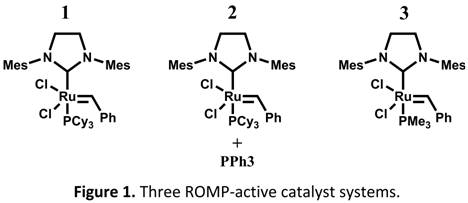
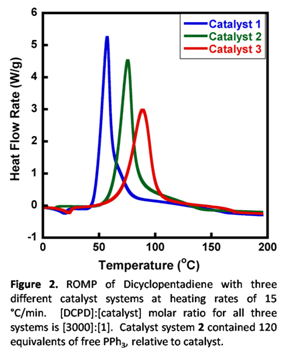
Our first approach to optimize healing capability entailed catalyst modification. Previous reports have shown that rapid polymerization kinetics are ideal in a self-healing composite, but if the reaction's initiation kinetics are too rapid, the quality of the polymerized healing agent can decrease (e.g. due to limited time to wet the crack surface and dissolve the catalyst produces polymerized healing agents with weaker adhesive and cohesive strengths). Therefore, a catalyst whose latency (i.e. bulk initiation rate) can be fine-tuned to meet certain substrate wetting time and catalyst dissolution requirements, without sacrificing rapid bulk polymerization propagation kinetics, is preferred. With this in mind, we developed two novel catalyst systems whose initiation kinetics (relative to Grubbs' catalyst) can be fine-tuned to a desired level, but still matches the superior propagation polymerization kinetics of Grubbs' catalyst. The Grubbs' 2nd generation catalyst and the two catalysts developed in this study are shown in Figure 1. The bulk initiation rate of the three catalyst systems can be determined in a differential scanning calorimeter (DSC) by comparing the temperature at which the exothermic reaction occurs, with higher temperatures correlating to increased catalyst latency. As seen in Figure 2, catalyst system 2 showed increased latency when polymerizing DCPD over the 2nd generation Grubbs' catalyst (catalyst 1 in the figure 1), followed by catalyst 3, which showed superior latency. In addition, it was observed that adding increasing amounts of PPh3 to catalyst system 2 acted to further fine-tune its latency. Hence, by using combinations of catalyst systems 2 and 3, virtually any desired degree of latency can be achieved.
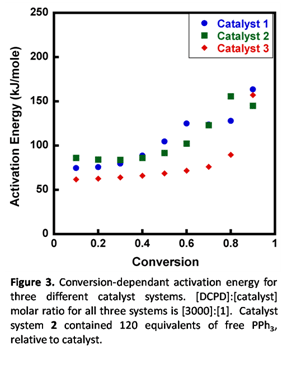
The ability to fine-tune bulk initiation rates should, ideally, not come at the cost of reducing polymerization propagation kinetics. Figure 3 shows the conversion-dependant activation energy of all three catalyst systems. As can be seen from the activation energy at low conversions (which is most representative of propagation kinetics), all three catalyst systems result in comparable polymerization, suggesting that there is no real opportunity cost to tuning the latency with our catalyst systems.
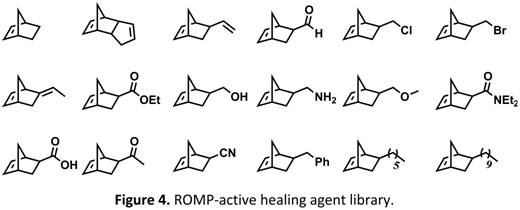
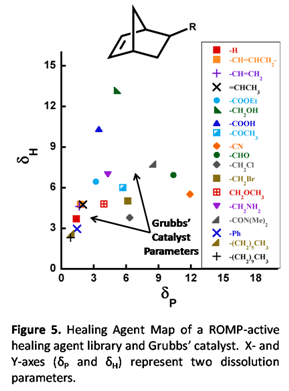
Additionally, we developed various different ROMP-active healing agents, screened their ability to self-heal, and use our experimental results to develop models that can be used to predict the performance of other healing agents. The healing agents used here are shown in Figure 4, all of which were either purchased or synthesized via literature preparations. As discussed earlier, rapid polymerization kinetics are ideal so long as the healing agent has sufficient time to first dissolve an appreciable amount of catalyst particles present on the crack plane of a self-healing polymer. Thus, a model was developed to make predictions regarding the dissolution rate of the Grubbs' catalyst in our small healing agent library. This model was constructed by experimentally determining sets of dissolution parameters for each of the healing agents and the catalyst. Detailed information on the calculation of these dissolution parameters can be read elsewhere [[3]]. These parameters are plotted on a Healing Agent Map (Figure 5) where each [x,y] coordinates represents two of the dissolution parameters for a given chemical. With this information, liquids (i.e. healing agents) should dissolve solids (i.e. Grubbs' catalyst) with similar dissolution parameters (in other words, liquids and solids within a close proximity to each other on the Healing Agent Map). This was experimentally shown to be the case. Perhaps even more useful, it was shown that blends of healing agents take on dissolution parameters intermediate between the individual components of the blend, and blends with dissolution parameters approximately identical to the catalyst were shown to exhibit superior dissolution rates.
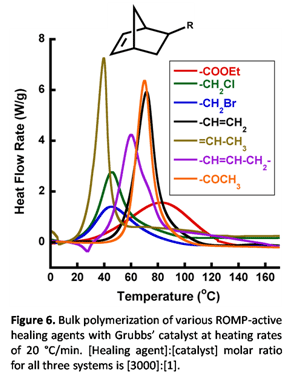
Finally, since fast polymerization kinetics are desired to self-heal in a reasonable time period, bulk reaction kinetics of several of the healing agents in Figure 4 with the Grubbs' catalyst were evaluated. Polymerization kinetics of several healing agents could not be evaluated in the bulk for various reasons (e.g. too slow reaction, too fast reaction, non-liquid healing agents), but those that could be evaluated were monitored by DSC, shown in Figure 6.