Reports: G7
47999-G7 Hierarchically-Assembled Segmented Polyurethanes: Mechanical Reinforcement in a Multi-Phase Elastomeric System
An examination of natural materialshave revealed that a key design component is a hierarchical architecture that, under deformation or stress, can utilize the multiple levels of organization to absorb energy and reinforce the material such that it will not permanently deform even in complex environments. Bone, an inorganic/organic composite, is made up of inorganic plate-like blocks (hydroxyapatite) encapsulated by an organic matrix composed of type I collagen1-3. Similarly, nacre has a brick and mortar type configuration with bricks of crystallized inorganic minerals (aragonite) mortared together with a complex mixture of proteins4,5. Spider silk, a material with a tensile strength superior to high-grade steel, is a natural thermoplastic elastomer composed of alanine-rich blocks of highly crystalline beta-sheet domains and an "oriented" amorphous, glycine-rich continuous matrix6-8.
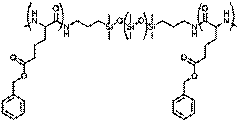
Figure 1 - Poly(benzyl-L-glutamate)-b-poly(dimethysiloxane)-b-poly(benzyl-L-glutamate)
The goal of this research is to design hierarchically-assembled segmented polyurethanes with enhanced mechanical function by incorporating liquid crystalline units within the soft phase, which is motivated by recent enhancements in toughness through the introduction of crystalline units in the continuous domain of polyurethanes. Segmented polyurethanes (PUs) are multi-block polymers usually consisting of an amorphous, flexible soft segment and a rigid crystalline or high glass transition temperature (Tg) hard segment. The flexible segments form a continuous domain, which is anchored by physical crosslinking of the hard segment aggregates. The dynamic framework of PUs offers many adjustable routes, namely the ability to modify the constituents of the hard and soft domains.. In order to enhance the mechanical properties, we propose to modify a poly(dimethysiloxane) (PDMS) soft segment by incorporating a polypeptide, poly(benzyl-L-glutamate) (PBLG), that is known to fold into defined secondary structures (alpha-helix, beta-sheet) at the PDMS terminus (Figure 1). We expect that the peptidic ordering within the soft block will provide additional energy absorption.
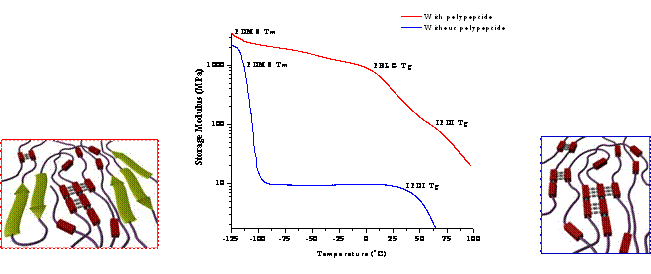
Figure 2 Comparison of dynamic storage modulus of segmented polyurethanes with an amorphous hard segment (IPDI-BDO) and either a peptidic or non-peptidic soft segment. Schematic: Left - peptidic PU; Right- non-peptidic PU
We have successfully synthesized the desired peptidic soft segment (~5000 g/mol, ~10 PBLG units, polydispersity <1.1) through a method devised by Deming et al.9,10 and incorporated the peptidic soft segment into two polyurethanes (limited solubility for molecular weight analysis); one containing a crystalline hard domain (hexamethylene diisocyanate/1,4 butanediol; HDI-BDO) and the other a non-crystalline, high Tg hard domain (isophorone diisocyanate/1,4 butanediol; IPDI-BDO). Preliminary thermal and mechanical results of solution cast films (THF at elevated temperatures) of the non-crystalline hard domain polyurethane with/without the peptidic soft segment indicate that the peptidic copolymers promote an increase in mechanical properties (Figure 2; Table 1). These materials are also melt-processable (Figure 3).
In Figure 2, it is seen that the non-peptide based PU loses significant mechanical properties around -115 ºC (melting temperature of PDMS), and flows at 65 ºC (Tg of IPDI-BDO), while the PU containing the peptide segment does retains its mechanical integrity until 15 ºC (PBLG Tg). Through the addition of the polypeptide segments, the initial modulus has been improved by an order of magnitude, 0.141 MPa to 2.5 MPa, but, as expected, a reduction in extensibility is observed (Table 1). Despite the significant decrease in elongation-at-break, their potential as impact/scratch resistant films/coatings is highlighted by the enhancement in toughness at 10% strain, which was dramatically increased from 6.4 MJ/m3 to 43.9 MJ/m3.

Figure 3 Melt-pressed IPDI-BDO-(PBLG-b-PDMS-b-PBLG). Conditions: 125 °C; 20,000 psi; pre-heated for 3 minutes; loaded 5 minutes.
Table 1 - Tensile properties of segmented polyurethanes with and without peptidic soft segment
|
Polyurethane (IPDI-BDO) |
Initial Modulus (MPa) |
Ultimate Tensile Strength (MPa) |
Toughness at 10% strain (MJ/m3) |
Elongation-at-break (%) |
|
Without polypeptide |
0.141 ± 0.01 |
4.2 ± 0.02 |
6.4 ± 0.30 |
110.6 ± 6.4 |
|
With polypeptide |
2.5 ± 0.5 |
4.75 ± 1.2 |
43.9 ± 18.4 |
15.3 ± 9.1 |
An additional mode of mechanical reinforcement and control of secondary structure in these new energy-absorbing polyurethane elastomers was also explored by the SUMR scholar via blend studies of the peptidic soft segment and/or PBLG-coated POSS11 (polyhedral oligomeric silsesquioxanes). It was anticipated that these PBLG-tethered POSS will preferentially associate with the soft phase and modulate the secondary structure of PBLG (Figure 4) without significant loss of hard domain microstructure when incorporated in the PU. Initial observations of blends PBLG /PBLG-tethered POSS blends suggest that the changes in secondary confirmation are highly correlated to repeat length of PBLG compared to the PBLG tether length, necessitating controlled PBLG growth from the functionalized POSS.
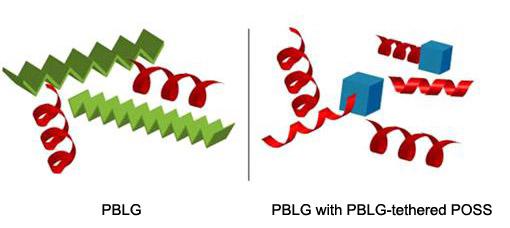
Figure 4 Schematic of POSS-PBLG interactions
The main focus of the second year is to understand the microstructure and deformation mechanics of the peptidic PUs. Initial SAXS data suggest a microphase-separated morphology, with the non-peptidic PU having an average domain spacing of 11 nm and the peptidic PU having a domain spacing of 23 nm. However, in order to understand how the peptide units contributes to the enhanced thermal and mechanical behavior of the polymer, dynamic (under deformation) small-angle and wide-angle X-ray scattering studies will be conducted. These studies, coupled with extensive tensile profiles, will allow the development of a structure-property profile for high performance materials with tailored mechanical responses.
References
[1] Weiner, S; Wagner, H. D.; Annu. Rev. Mater. Sci., 1998, 28, 271-298
[2] Ortiz, C.; Tai, K.; Dao, M.; Suresh, S.; Palazoglu, A.; Nature Materials, 2007, 6, 454-462
[3] Gauckler, L. J.; Studart, A. R.; Bonderer, L. J.; Science, 2008, 319, 1069-1073
[4] Bruet, B. J. F.; Boyce, M. C.; Qi, H. J.; Oritz, C.; Frick, L.; Tai, K.; Panas, R; J. Mater. Res, 2005, 20, 2400-2419
[5] Weiner, S.; Addadi, L.; J. Mater. Chem, 1997, 7, 689-702
[6] Knight, D. P.; Vollrath, F.; Nature, 2001, 410, 541-548
[7] Grubb, D. T.; Jelinski, L. W.; Macromolecules, 1997, 30, 2860-2867
[8] Jelinski, L. W.; Simmons, A.; Ray, Ed.; Macromolecules, 1994, 27, 5235-5237
[9] Deming, T. J.; Brzezinska, K. R.; Macromolecules, 2001, 34, 4348-4354
[10] Deming, T. J.; Brzezinska, K. R.; Curtin, S. A; Macromolecules, 2002, 35, 2970-2976
[11] Kuo, S-W.; Lee, H-F.; Huang, W-J.; Jeong, K-U.; Chang, F-C.; Macromolecules, 2009, 42, 1619-1626.



