Reports: AC2
46636-AC2 Quantum Mechanical Calculation of Hydrogen Isotope Exchange Thermodynamics and Kinetics on Petroleum Model Compounds
The second year of research into the thermodynamics and kinetics of H-D isotopic exchange on kerogen-related molecules was conducted by performing calculations on H abstraction reactions, H-D exchange with H2O, SiOH and AlOH groups, and H-D frequency analyses on kerogen components other than alkanes. This research will be used to help interpret observed H-isotope fractionation factors in ancient organic matter from sedimentary rocks. Theoretical values are needed because molecular-specific isotopic analyses have recently been made possible, but experimental values are difficult to obtain especially for specific sites within compounds. Observed fractionation trends will require knowledge of both the equilbrium fractionation factors and exchange rates as a function of temperature.
The B3LYP/6-311++G(d,p) method was used in all cases and was compared with CBS-Q and MP2 results to verify accuracy. Calculated frequencies were used to estimate ln(b) values for alkane compounds. The α values were then calculated by the difference between the alkane ln(b) values and those of HDO, D2O and CH3D. The calculated ln(b) value is in reasonable agreement with a previously derived ln(β) for water vapor. A test suite of compounds (CH4, C2H6, (CH3)2CH2, C6H12 and (CH3)3CH) were used to determine the level of agreement between empirically-derived and quantum mechanical α values. The results predict that the preference for D in these alkanes should be in the order of tertiary > secondary > primary C-H groups. The tertiary and secondary C-H groups have predicted fractionation factors > 1 with respect to HDO at 25°C, but these a values drop considerably with increasing temperature such that all a values are < 1 by 100°C. Around 225°C, the a values for all sites converge towards a single value, so D would not discriminate among sites above this temperature.
Similar calculations on the test suite of compounds listed in the Table 1 show a similar range of a-values as found for alkanes with some sites favoring D over the H site in H2O. The activation energy barriers calculated for H-D exchange with H2O (Figure 1) are significantly lower in this suite of compounds compared to the alkanes indicating that equilibrium will be reached sooner and at lower temperatures for these O-, N- and S-containing compounds. The energy barriers for exchange with Al-OH and Si-OH groups were lower still suggesting that H-isotope exchange with mineral surfaces will be a key mechanism.
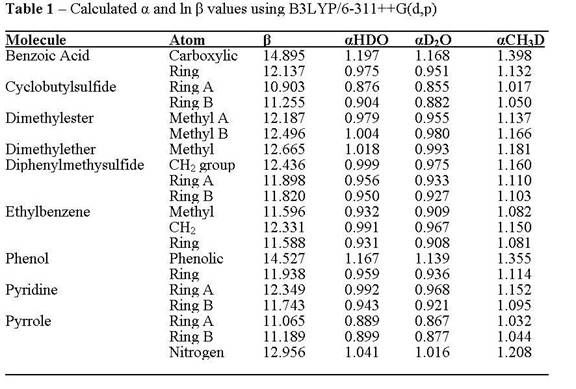
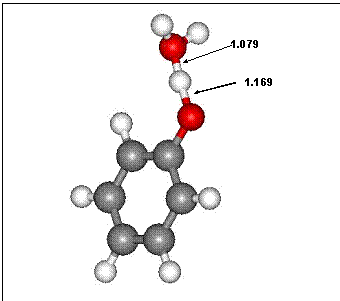
Figure 1 Model of exchange reaction between phenol and H2O. The steps along the H---O transfer path were constrained at every 0.1 Å allowing the remaining atoms in the cluster to relax in order to find a minimum energy pathway between the reactants and products. Barrier heights were estimated for both the ionic and radical electronic states of the system, but the ionic state was significantly lower in energy for all pathways.
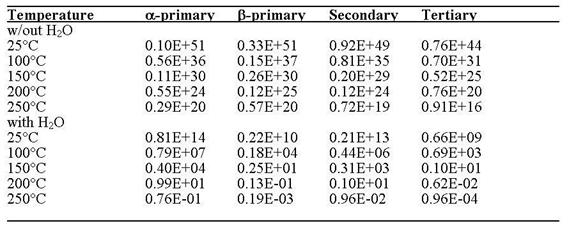
When H2O is present, the activation energy barriers and half-lives are lower (Table 2). Using the rates predicted by B3LYP/6-311++G(d,p), for the a-primary site at 100°C, the half-live is approximately 8 million years. Approaching equilibrium is generally considered to require about 5 half-lives, so one could not expect to be able to use calculated a-values unless the sample had been exposed to H2O at 100°C for around 40 million years. The calculated half-lives for the b-primary sites are significantly lower. For example, the t1/2 for the b primary carbon at 100°C is on the order of 2000 years (Table 2). The large difference in calculated rates for the a- and b-primary sites implies that exchange at the latter methyl groups may be significant or even reach equilibrium whereas the former may undergo no or insignificant H-D exchange. Secondary sites are predicted to exchange at a rate intermediate between the a and b primary sites. Tertiary sites have the most rapid exchange rates (Table 2) with a t1/2 around 700 years at 100°C. As the temperatures rise to 200°C, the half-lives generally are on the order of a year or less. One could expect equilibrium to be reached at these sites and the calculated a-values calculated could be applied.
Table 2 Estimated half-lives (t1/2) in years for B3LYP/6-311++G(d,p) H abstraction reactions on 2-methyl butane with and without H2O present.