Reports: AC1
46011-AC1 Supramolecular Synthesis of Molecular Co-crystals: A Versatile Approach to Modulating Physical Properties of Functional Molecular Solids
1. INTRODUCTION
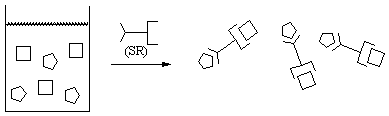
|
In order to develop versatile supramolecular synthetic strategies that can be applied in the hydrogen-bond (HB) driven assembly of co-crystals it is necessary to identify building blocks that display reliable binding preferences in the presence of a range of chemical functionalities. In this project we have taken significant steps towards mapping out supramolecular structural preferences of common chemical functionalities by examining the competition and balance between non-covalent interactions. We established that it is possible to manipulate the way in which molecules recognize and bind to each other by altering the strengths of site-specific complementary hydrogen bonds. We have realized this goal by allowing a library of custom-designed probe molecules (with multiple HB donors/acceptors) to react with tagerget molecules in order to identify a ranking of synthons within an intermolecularly competitive framework. The design of the probe molecules, as well as of the observed binding preferences, have been rationalized using an electrostatic view of intermolecular interactions, Scheme 1.1.
Scheme 1.1 A one-pot supramolecular reaction between two different
targets and a ditopic supramolecular probe resulting in 1:1:1 ternary
supermolecules.
2. RESULTS
2.1 New hydrogen-bond donors
A synthetic-structural study[1] of co-crystals of N-heterocycles and cyanooximes furnished information that allowed us to draw some conclusions about the structural behavior of the latter functional group. There are four phenylcyanooximes in the CSD[2] and all of them contain OH/N (nitrile) hydrogen-bonded catemers. This is in contrast to the structural preference of aromatic aldoxime and acetyloximes which show a preference for dimers that utilize the OH moiety as a donor, and the oxime nitrogen atom as the hydrogen-bond acceptor. 25 out of 32 structures show this arrangement and seven of 32 structures contain OH/N(oxime) catemers. This change in pattern preference may come about because the nitrile moiety is a better hydrogen-bond acceptor, -224 kJ mol-1 (N-nitrile) vs. -125 kJ mol-1 (N-oxime) as indcated by AM1 calculated electrostatic potential, Scheme 2.1). This makes the cyanooxime hydrogen-bond donor more inclined to interact with the nitrile moiety, than with the oxime nitrogen atom, which is in accordance with the best-donor/best-acceptor guideline.
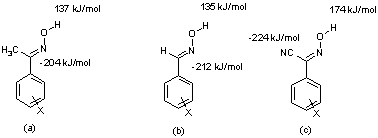
Scheme 2.1 Molecular electrostatic potential data (maxima and minima) for three generic oximes (a) acetyloximes, (b) aldoximes, and (c) cyanoximes.
We have shown that cyanooximes have considerable potential as effective co-crystallizing agents. As single-point hydrogen-bond donors they should be comparable to (if not better than) carboxylic acids, they are synthetically readily available, and they also tend to be soluble in a wide range of solvents, which facilitates solution-based co-crystal synthesis.
2.2 Combining hydrogen bonds and halogen bonds in supramolecular assembly
Hydrogen bonds and halogen bonds play important roles in the synthesis of co-crystals,[3] and the combination of both in a unified synthetic strategy is particularly attractive as this may allow for the construction of more complex assemblies composed of a larger number of different molecular species. By combining a halogen-bond donor with an appropriate molecule containing (a) an effective halogen-bond acceptor and (b) a self-complementary hydrogen-bonding moiety our synthetic strategy is clearly defined.[4] Two examples of co-crystal synthesis using 1,4-diiodotetrafluorobenzene A, as one of the building blocks, and a pyridine-based molecule equipped with a self-complementary hydrogen-bond functionality are described briefly, Scheme 2.2.
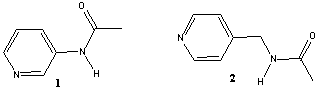
Scheme 2.2 Two building blocks for combined hydrogen-bond and halogen-bond based crystal engineering.
The crystal structure determinations of both A1 and A2 provided encouraging evidence that our synthetic plan had worked. The 1:2 stoichiometry in A1 and A2 is a consequence of the combination of a ditopic halogen-bond donor with two monotopic halogen-bond acceptor. The self-complementary N-H×××O=C hydrogen bond is also in place which means that the two components in this co-crystal are organized into 2-D sheets. There are no short contacts between molecules of A in this structure, Figure 2.1.
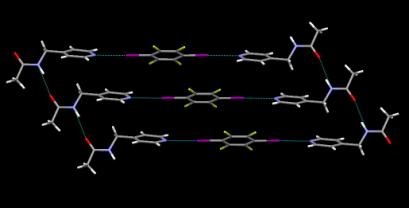
Figure 2.1 Hydrogen bonds and halogen bonds combine to create an infinite ladder in the crystal structure of A2.
So far, the primary intermolecular interactions have materialized as intended, and the synthetic strategy has worked to the point were we have maintained control over stoichiometry and, broadly speaking, the main supramolecular motifs in the two co-crystals A1 and A2.
2.3 Towards molecular capsules
The Pd-catalyzed Suzuki-Miyaura cross-coupling reaction is a well-established route for carbon-carbon bond formation and we have now synthesized[5] a tetraboronic pinacolyl ester functionalized cavitand, which allows for the preparation of a larger variety of functionalized deep-well cavitands or dimeric molecular capsules, Scheme 3.1.
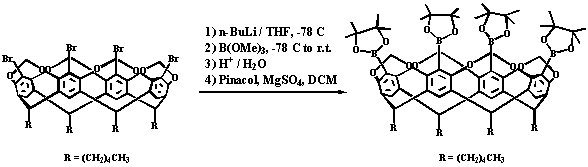
Scheme 3.1 Synthesis of tetraboronic pinacolyl ester functionalized cavitand.
We have successfully prepared a small library of functionalized cavitands and we have also been able to assemble them into a variety of supramolecular structures including polymers as well as the desired discrete capsules, Fig. 3.1.[6]
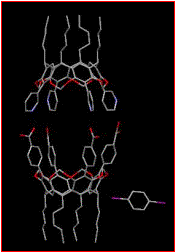
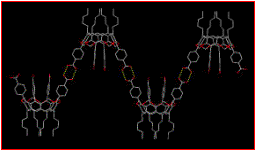
Figure 3.1. A supramolecular polymer of cavitands (left) and a discrete molecular capsule held together by -COOH×××N(py) hydrogen bonds (right).
The primary goal of this proposal was not to provide a detailed roadmap for the manufacturing of a device or material with precisely defined properties or performance. It has, however, produced several hypotheses-driven sets of experimental data that collectively offer the basis for a versatile synthetic strategy for the construction of molecular solids of considerable complexity and improved physical properties.
References
[1] Aakeröy, C.B.; Desper, J.; Salmon, D.J., Smith, M.M. CrystEngComm, 2009, 11, 439-433.
[2] Allen, F.H. Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380.
[3] Aakeröy, C.B.; Desper, J.; Fasulo, M.; Hussain, I.; Levin, B.; Schultheiss, N. CrystEngComm, 2008, 10, 1816-1821; Aakeröy, C.B.; Schultheiss, N.; Rajbanshi, A.; Desper, J.; Moore, C., Cryst. Growth Des., 2009, 9, 432-441.
[4] Aakeröy, C.B.; Scott., B.M.T.; Smith, M.M.; Urbina, J.F.; Desper, J., Inorg. Chem., 2009, 48, 4052-4061; Aakeröy, C.B., Hussain, I., Forbes, S., Desper, J.,Aus. J. Chem., 2009, 62, 899-908.
[5] Aakeröy, C.B., Schultheiss, N., Desper, J., unpublished results.
[6] Aakeröy, C.B., Chopade, P., Rajbanshi, A., Desper, J., unpublished results.



