Reports: B3
47113-B3 Electrochemical Activation of Cytochrome P450
 During the second
funding period of our project we continued to examine the factors required for in
vitro activation of bacterial Cytochrome
P450 from Bacillus megaterium
(P450 BM3). Following our previous
work, we engineered and expressed four different BM3 heme-domain mutants that
featured a surface cysteine (Cys387, Cys383, Cys62,
and Cys97), then labeled the resulting proteins with the
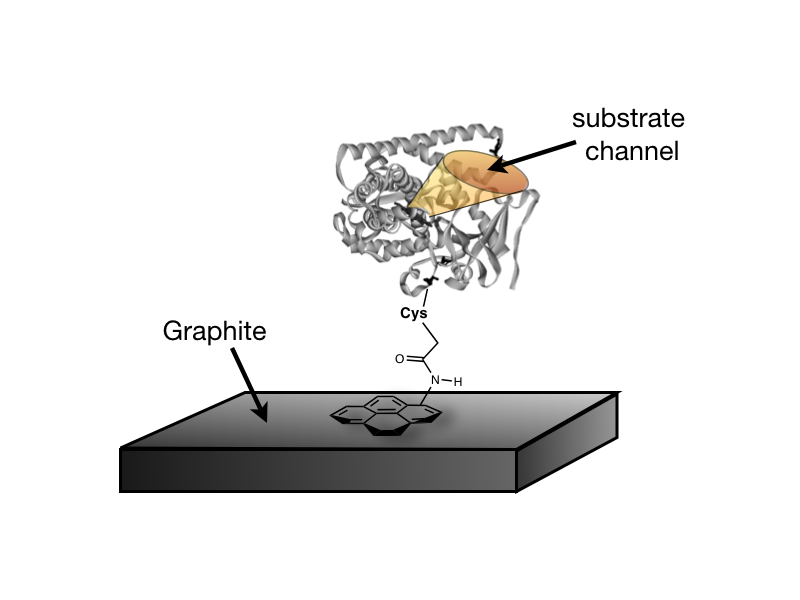
thiol-specific N-(1-pyrene)-iodoacetamide reagent. As illustrated, pyrene adsorbs to basel-plane graphite
immobilizing each of the heme domains onto the surface while keeping their
substrate-access channels exposed.
Respective Cys-heme distances for these conjugates are 19 , 26 , 19 ,
and 17 .
During the second
funding period of our project we continued to examine the factors required for in
vitro activation of bacterial Cytochrome
P450 from Bacillus megaterium
(P450 BM3). Following our previous
work, we engineered and expressed four different BM3 heme-domain mutants that
featured a surface cysteine (Cys387, Cys383, Cys62,
and Cys97), then labeled the resulting proteins with the
thiol-specific N-(1-pyrene)-iodoacetamide reagent. As illustrated, pyrene adsorbs to basel-plane graphite
immobilizing each of the heme domains onto the surface while keeping their
substrate-access channels exposed.
Respective Cys-heme distances for these conjugates are 19 , 26 , 19 ,
and 17 .
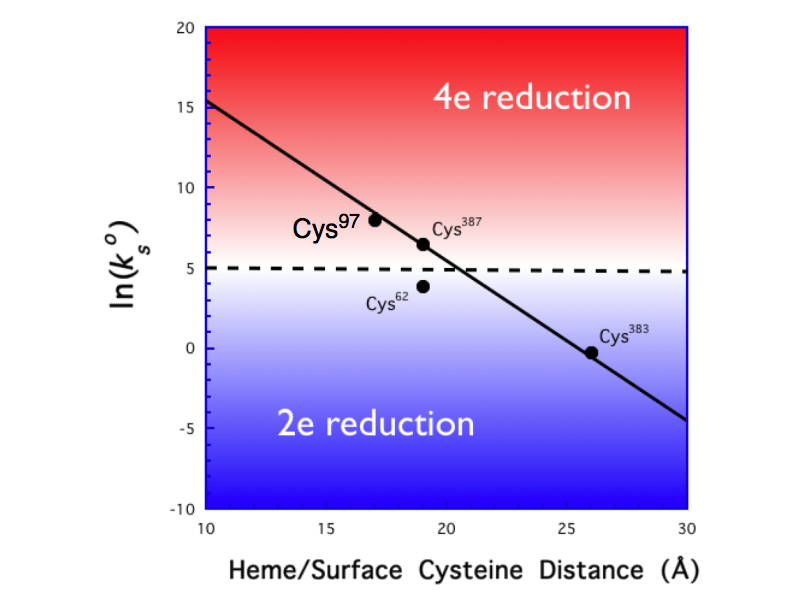
Using variable scan-rate cyclic voltammetry at a microelectrode, we have now measured the heterogeneous electron-transfer (ET) rate for the Fe(III/II) couple of electrode-bound Cys97 (ko= 2(1)x103 s-1) to go along with our previous measurements of the analogous rates of Cys387, Cys383, and Cys62. The rate is consistent with our previous data, falling on a ln(kET) vs. distance line drawn assuming a decay constant, b, of 1 -1.
Each of the bound proteins reacts catalytically with dioxygen upon reduction at the electrode surface. In the native system, ET into the Fe(III) heme of P450 from a reductase domain gives Fe(II), which binds dioxygen. A second proton-coupled reduction yields the Fe(III)-OOH hydroperoxy complex, which is protonated to give water and a high valent iron-oxo species known as compound I. Compound I is the catalytic species that initiates C-H bond activation via oxygen-atom transfer to produce an alcohol and to regenerate the resting Fe(III) heme. We had already established that Cys387 catalyzes the 4e- reduction of dioxygen to water without affecting any substrate oxidation: evidently the ET pathway is too fast resulting in further reduction of the two-electron-reduced ferric hydroperoxy species before conversion to compound I. We have now carried out electrochemical experiments at a rotated-disk electrode (RDE) to determine the products of dioxygen reduction catalyzed by the other mutants. The diffusion-convection-limited current at an RDE depends on the angular rotational velocity (w s-1), the number of electrons transferred (n), and the bulk concentration of substrate (O2) according to the Levich equation:
IL = 0.62nFA2/3[O2]n-1/6w1/2
(where F is Faraday's constant, A is the electrode area—determined using ferrocene as a calibration standard, and n is the kinematic viscosity of the electrolyte solution). Plots of IL vs. w1/2 are linear up to rotation rates of 1600 rpm for all of the mutants studied. Based on the slopes of the lines, we calculate n values of 1.9(2) for Cys383, 2.6(2) for Cys62, and 4.0(2) for Cys97. An n = 2 value implies reduction by 2e- to peroxide, whereas n = 4 indicates reduction by 4e- to water; the 2.6 value found for Cys62 suggests that dioxygen is catalytically reduced to both peroxide and water, in roughly a 3:2 ratio.
 These data (see plot) demonstrate that a
heterogeneous ET rate on the order of ~10s-1 appears to lie at the
dividing line between 2e- vs. 4e- dioxygen reactivity for this system. We believe that substrate turnover may
be possible only for systems that activate dioxygen by 2e-, thereby
avoiding uncontrolled electron transfer into the heme center that reduces
Fe(III)-OOH by two additional electrons, thereby prohibiting formation of
compund I.
These data (see plot) demonstrate that a
heterogeneous ET rate on the order of ~10s-1 appears to lie at the
dividing line between 2e- vs. 4e- dioxygen reactivity for this system. We believe that substrate turnover may
be possible only for systems that activate dioxygen by 2e-, thereby
avoiding uncontrolled electron transfer into the heme center that reduces
Fe(III)-OOH by two additional electrons, thereby prohibiting formation of
compund I.
To test this hypothesis, we have initiated substrate turnover experiments via bulk electrolysis of bound P450s in the presence of dioxygen and styrene. Large-surface-area carbon-felt electrodes were modified with pyrene-labeled P450s, then held at potentials just cathodic of the Fe(III/II) heme couple in phosphate buffer solution that was 1mM in styrene. After two hours, samples were removed and analyzed by GC-MS. Neither the Cys387 nor Cys97 systems showed detectable amounts of oxidized styrene, while both Cys62 and Cys383 showed the presence of styrene oxide and benzaldehyde. Addition of catalase to the reaction mechanism had no effect on the catalytic turnover, implying that the reaction proceeded through the native mechanism and not the peroxide shunt. We are currently working out assays to quantify this reaction (initial results based on coulometry suggest ~ 5,000 turnovers), as well as to examine the protein-film reactivity towards other substrates (e.g., lauric acid).



