Reports: G4
48060-G4 Intramolecular and Intermolecular Kinetic Isotope Effects for Highly Stereoselective Oxazaborolidine- Catalyzed Reductions
Our laboratory has been involved in the development of new methodologies for the investigation of asymmetric reactions. The Corey-Bakshi-Shibata (CBS) reduction utilizes an oxazaborolidine catalyst derived from proline. The most commonly used variant of the CBS catalyst, Me-CBS, has a tremendously broad substrate range. We continue to apply our methodologies toward understanding the simulataneous substrate range and high stereoselectivity observed with this catalyst. Our recent findings suggest that the CBS catalyst has two modes of enforcing stereoselection. It appears that stereoselection is mediated by the avoidance of steric interaction between the large (aryl) substituent and the prolinol substituents on the catalyst with aralkyl ketone substrates that have no α-substituent. Our current hypothesis is that the transition state that describes this process possesses a chair-like conformation of the active atoms. By contrast, computed transition structures and experimentally-determined 2H and 13C kinetic isotope effects (KIEs) suggest that stereoselection is mediated by the avoidance of coincidence between the large (aryl) substituent and the B-methyl group. The transition structure computed for the Me-CBS-catalyzed reduction of 2',5'-dimethylisobutyrophenone appears to possess a boat-like arrangement of the atoms that participate in bond breaking and bond formation (Fig. 1).
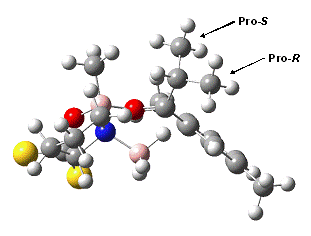
Figure 1. Computed transition structure [B3LYP/6-31+G(d,p)]for Re-attack of hydride upon 2',5'-dimethylphenyl isopropyl ketone. The two phenyl groups on the CBS catalyst are denoted by gold spheres. At center is the boat-like arrangement of atoms that participate in bond breaking and bond making.
Our initial foray into this research problem utilized a new methodology developed in our laboratory to measure 13C KIEs at enantiotopic groups. The expectation was that groups that experienced significant steric occlusion in the transition state would possess inverse 13C KIEs as the result of a tighter CH stretch vibrations. Surprisingly, we found that there were no significant 13C KIEs at either enantiotopic group. The only significant 13C KIE was found to reside at the carbonyl position (Fig. 2A). We found that 13C KIEs computed from the boat-like transition state agreed with our experimentally-determined KIEs (Fig. 2B).

Figure 2. (A) Experimental 13C KIEs (k12C/k13C) for the (S)-Me-CBS catalyzed borane reduction of 2',5'-dimethylphenyl isopropyl ketone. (B) Computed KIEs based on the [B3LYP/6-31+G(d,p)] transition structure for Si attack.
Based upon the observed steric interactions in the computed transition structure for the Me-CBS-catalyzed borane reduction of 2',5'-dimethylisobutyrophenone, we were surprised to have not seen an observable 13C KIE (Fig. 3). We employed our new methodology for measuring 2H KIEs at enantiotopic groups to see if the steric interactions suggested by the transition structure would yield an inverse 2H KIE at either or both enantiotopic methyl groups. This methodology employs two competition reactions to arrive at the 2H KIEs at each prochiral methyl group. The first competition reaction (Scheme 1A) involves competition between the perprotiated isotopolog (1) and the isotopolog labeled at both prochiral methyl groups (2). This competition reaction yields the product of the KIEs at the prochiral methyl groups, assuming the rule of the geometric mean (eq 1). The second competition reaction involves competition within a racemic mixture of isotopomers in which one prochiral methyl group is labeled (Scheme 1B). This competition reaction allows for the measurement of the ratio of 2H KIEs at each prochiral methyl group (eq. 2). From these two measured rate ratios, the 2H KIEs at each prochiral group can be computed (eqs. 3a&3b).
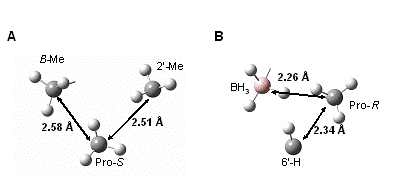
Figure 3. Nearest neighbor non-bonding interactions in the computed transition structure for the A) pro-S and B) pro-R methyl groups. Distances are nearest HH contact distances between the groups indicated.
Scheme 1. The two competition reactions by which 2H KIEs at enantiotopic methyl groups are determined.
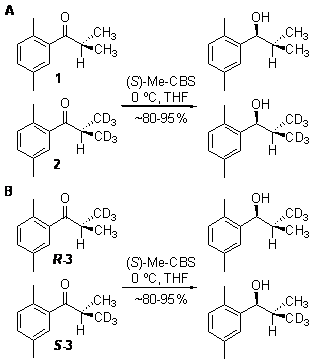
![]()
![]()
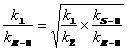
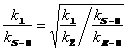
Our results from the measurements described in Scheme 1 resulted in values that are commensurate with what might be expected, given the interactions outlined in Fig. 3. The pro-R methyl group has closer HH approach distances than the pro-S methyl group. These interactions are reflected in the larger inverse 2H KIE measured for the pro-R methyl group (Fig. 4). Having observed a measurable 2H KIE at both enantiotopic methyl groups but having observed little or no effect in the 13C KIEs suggests that there may be a compensatory interaction at the carbon centers that attenuates the expected inverse 13C KIE. Our current hypothesis is that the polarization that occurs in the transition state near the carbonyl may weaken C C bonds that contain the prochiral methyl groups. This interaction might explain the lack of substantial 13C KIEs at the prochiral groups.
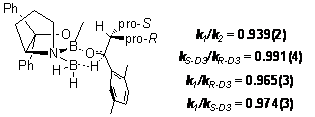
Figure 4. Relative rate constants computed from fractionation in reisolated reactant and resulting KIEs upon the prochiral methyl groups in the substrate. Errors in the last digit are in parentheses.
We continue to explore the nature of stereoselection and steric interactions in the CBS reaction. We are currently exploring the CBS reduction of ynones, where the isopropyl probe group is the large' substituent. These reactions allow us to explore steric interactions in an environment where one might expect little steric presence. Likewise, we will be studying these reactions in the context of Alpine-Borane reductions. This comparison is essential, since the CBS reduction is not highly selective for the reduction of ynones unless super-stoichiometric amounts of the catalyst are employed. By contrast, Alpine-Borane has been shown to reduce ynones with high selectivity. A more refined understanding of these reactions using a combined computational and experimental approach promises to elucidate the interactions that are responsible for stereoselectivity in these stereoselective reductions. These findings may allow for the rational design of a better CBS catalyst for the reduction of ynones. Broadly speaking, a better understanding of the process of stereoselection, in general, promises to lead to the development of better asymmetric catalysts. Improved methods for asymmetric catalysis promise to reduce the waste that is currently associated with most industrial scale processes used to synthesize molecules with stereogenic centers. In turn, these developments promise to promote the more efficient use of petroleum feedstocks as the conservation of these precious resources becomes more critical.



