Reports: G3
46939-G3 Toward Understanding Biological Nitrogen Fixation: Direct Electrochemical Studies of Nitrogenase
After four decades of extensive research, mechanistic details of how nitrogenase accomplishes the conversion of dinitrogen into ammonia still remain a mystery. Reduction of the nitrogenase active site metal cluster, FeMoco, is postulated to be required for substrate binding and has only been achieved under turnover conditions in solution. Consequently, it has been challenging to populate substrate/intermediate-bound forms of nitrogenase in sufficient quantities for physical characterization. In order to gain a better understanding of electron transfer processes within nitrogenase, we had proposed to develop methods for the direct interrogation of the redox properties of the Fe-S clusters situated in both nitrogenase components, the Fe-protein (FeP) and the MoFe-protein (MoFeP). The financial support by the ACS Petroleum Research Fund has enabled me and my group to pursue this very high risk/payoff line of research for the last two years. Specifically, these funds were primarily used for supporting a graduate student, Lauren Roth during the first year of funding and an undergraduate researcher, Thomas Ni, during the second.
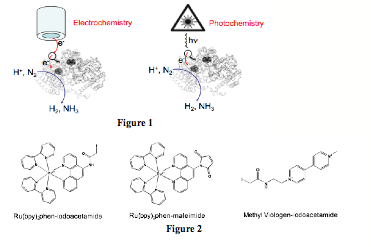
Despite the obvious need to establish the redox properties of the nitrogenase Fe-S clusters and to electronically access them, there are no published reports on their direct electrochemistry and redox activation, possibly owing to their burial within the protein medium, particularly in the case of MoFeP. Thus, during the second year fo ACS PRF-G funding, we continued on our efforts that focused on the site-selective labeling of MoFeP with redox- or photo-active functionalities that would provide conduits for rapid electron transfer to the buried Fe-S cluster, FeMoco (Fig1). With this goal in mind, we prepared Cys-specific methyl viologen (MV) and Ru-polypyridine derivatives that could be utilized for electrochemistry and photochemistry experiments, respectively (Fig2).
During the last year, we concentrated our efforts on labeling three different MoFeP constructs that pose Cys-residues in different surface locations:
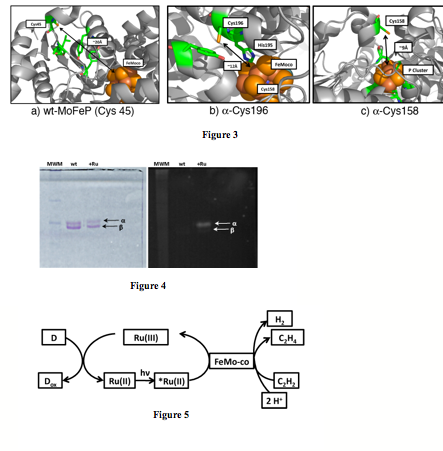
1) Wild-Type MoFeP (Fig3a): Wild-type MoFeP contains only a single surface Cys (a-Cys45) that is adequately solvent-exposed. Fortuitously, Cys45 is close enough to FeMoco (<20 Ang) to allow weak to moderate electronic coupling.
2) a-H196C MoFeP (Fig3b): The a-196 position is separated from FeMoco by only one intervening residue (a-His195) which is directly H-bonded to FeMoco. The distance between a-196 and FeMoco is only 12 Ang, which should provide for a very strong electronic coupling between the active site and the protein surface. This mutant also contains the native a-Cys45.
3) a-L158C MoFeP (Fig3c): The a-158 position lies right above the P-cluster (~ 9 Ang), which is postulated to be the electron relay site between FeP and FeMoco. Thus, the a-L158C MoFeP is the only construct whereby the electron transfer pathway is similar to the native enzyme (external donor -> P-cluster -> FeMoco). This mutant also contains the native a-Cys45.
As in the previous year, a significant fraction of the funding period was spent on the optimization of the protein labeling conditions and the determination of the location of functional groups on the protein surface. These experiments are challenging due to the large size (~250 kDa) of MoFeP, in addition to the difficulty in chromatographically separating the products from unlabeled protein. We have succeeded in efficiently labeling all three MoFeP constructs with the Cys-specific Ru(bpy)2(Phen-IA) derivative. Inductively-coupled optical emission spectroscopy (ICP-OES) and SDS-PAGE experiments indicate that each construct is labeled at the a-subunit (Fig4) and contains the correct number of labels per a-subunit. All labeled-constructs retain >50% of wild-type activity, indicating that labeling does not adversely affect MoFeP structure or compromise its Fe-S clusters. With this information in hand, we have been pursuing several strategies in parallel to determine of the exact location of Ru-labels on the a-subunit through protein digestion coupled with mass spectrometry (MS), and X-ray crystallography. We have had the most success with trpysin and cyanogen bromide (CNBR), which efficiently digest the MoFeP into fragmens that should be amenable to MS analysis. However, despite numerous attempts, which included the purification of individual peptide fragments, we have not been able to ascertain the location of Ru-functional groups on MoFeP with conventional MS techniques (MALDI, electrospray MS). We have recently started a collaboration with the Dorrestein Lab at UCSD, who are experts in the MS analysis of complex natural products. Using a comprehensive strategy that involves tandem mass spectroscopy and software developed for the MS analysis of posttranslationally modified peptides (InsPecT), we are hoping to soon pinpoint the location of Ru-photosensitizers on MoFeP surface. With the assumption that all three constructs have been Ru-labeled at the desired locations, we have started to examine whether MoFeP can be activated by light. Our initial experiments use a flash-quench scheme (Fig5), with dithionite, oxalate, or EDTA as sacrificial electron donors, and protons or acetylene as substrates (because their reduction only require two electrons). Promisingly, we observed significant amounts of ethylene formation from acetylene reduction when dithionite was used as a donor. Unfortunately, control experiments using only the Ru(bpy)2(Phen-IA) photosensitizer (in the absence of MoFeP) also produced ethylene. In all other cases, no significant hydrogen or ethylene production was observed. While the photochemical reduction of acetylene by Ru is in itself interesting (whose mechanism we are studying), these experiments show that alternative strategies have to be devised. Given that the a-C196 and a-C158 constructs both feature Ru-labels at a short enough distance for efficient electron transfer into the MoFeP conduit, there are several possible interpretations of our results:
1) The ATP-dependent interactions between FeP and MoFeP are absolutely necessary for MoFeP activity.
2) The photochemical scheme we utilize effects a one-electron, but not a two-electron reduction of MoFeP, explaining the lack of hydrogen or ethylene production.
3) The Ru-labels are located in unintended locations on the MoFeP surface.
Once our ongoing MS experiments ascertain the location of Ru-labels, we will be situated to probe by EPR spectroscopy whether FeMoco can be photochemically reduced by one electron. These experiments will determine the feasibility of two-electron photoreduction schemes, and provide clues as to whether interpretation 1 (the absolute necessity of FeP) is correct.