Reports: AC1
47810-AC1 Computational Design, Virtual Screening, and Experimental Validation of Chiral Catalysts
Reduction of acrylic acid derivatives such as acrylamides via catalytic enantioselective hydrogenation has several important applications in the synthesis of chiral a,b-unsaturated amides, esters and other carboxylic acid derivatives. The goal of the present research is to identify and/or design suitable chiral ligands in order to catalyze highly stereoselective reactions. So far, the number of good catalysts, in terms of yields and stereoselectivity, are few. Among the most promising catalysts are complexes of ferrocenylamine sulfides and Walphos type ligands. Starting from our laboratory's experience in stereoselective hydrogenation and a fruitful collaboration with the Norrby group, we have initiated a combined experimental and computation investigation to approach this problem of choosing and/or designing suitable catalysts.
Previous results of others for rhodium-catalyzed hydrogenation of acrylic acid derivatives indicated Walphos ligand 3 as having promising potential. Our follow-up study of rhodium-catalyzed hydrogenation with ligand 3 and acrylamides 1a and 1b is summarized in Table 1. The best results are shown in entry 14 whereby a 72% e.e. was obtained.
|
|
|||||
|
entry |
substrate |
solvent |
temp/°C |
% conv |
e.e. |
|
1 |
1a |
MeOH |
r.t. |
100 |
69 |
|
2 |
1a |
MeOH |
0 |
100 |
69 |
|
3 |
1a |
MeOH |
-78 |
<5 |
54 |
|
4 |
1a |
MeOH |
50 |
100 |
69 |
|
5 |
1a |
CH2Cl2 |
r.t. |
100 |
67 |
|
6 |
1a |
THF |
r.t. |
100 |
50 |
|
7 |
1a |
toluene |
r.t. |
100 |
42 |
|
8 |
1a |
Et2O |
r.t. |
100 |
44 |
|
9 |
1a |
EtOH |
r.t. |
100 |
66 |
|
10 |
1a |
iPrOH |
r.t. |
100 |
63 |
|
11 |
1b |
MeOH |
r.t. |
100 |
64 |
|
12 |
1b |
MeOH |
50 |
100 |
65 |
|
13 |
1b |
CH2Cl2 |
r.t. |
100 |
71 |
|
14 |
1b |
CH2Cl2 |
0 |
100 |
72 |
|
15 |
1b |
CH2Cl2 |
-20 |
58 |
72 |
|
16 |
1b |
CH2Cl2 |
-40 |
<5 |
unk |
|
17 |
1b |
CH2Cl2 |
-78 |
<5 |
unk |
|
Table 1: Optimization studies for rhodium-catalyzed Hydrogenation; e.e. determined by GC. |
|||||
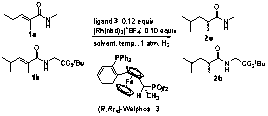
Based upon these results, we
applied the use of 3 to the
substrate 4 being studied in a
natural product synthesis. The best conditions for 4 resulted in a 95% yield and 85:15 d.r. for
product 5 (eq 1). Therefore, our experimental efforts
starting with the use of a previously developed ligand have provided only moderate
to good enantioselectivity, which provides an impetus for further catalyst ![]()
development.
As a fundamental basis for understanding catalyst stereoselectivity, we needed to probe the mechanism and energetics of the relevant hydrogenation reactions. In order to identify the rate determining and stereoselectivity determining steps of this reaction, we performed a density functional theory (DFT) study at the B3LYP level and characterized several of the hypothesized products and transition states. All calculations were performed using 6-31++G** basis set on non-metal atoms and LanL2TZ(f) basis set and ECP on the Rh atom. This study was based on the hypothesis that the basic mechanism of acrylamide hydrogenation is analogous to that of enamide hydrogenation and follows Scheme 1.
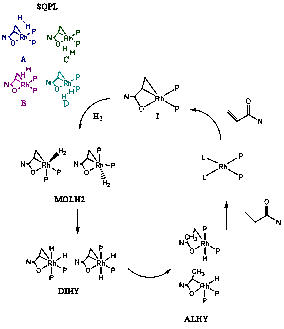
Scheme 1: Proposed mechanism for the Rh(I) catalyzed acrylamide hydrogenation. The four possible H2 approaches to the pre-catalyst I lead to four different pathways labeled as A, B, C and D in the paper.
Analogous to the results previously reported by Landis et al., the acrylamide hydrogenation starts with the approach of the H2 molecule to the square planar catalyst. For enamide hydrogenation, our starting catalyst model is formed by a Rh(I)(PH3)2(ethylenamide) complex (I) which interacts in four possible ways (A-D) with molecular H2. As shown in Scheme 1, H2 can approach from the top or from the bottom with respect to the plane defined by the four ligands. The energy profile of the reaction under study is depicted in Figure 1. In the discussion that follows, we report the most important energies for key species in kcal/mol (in parenthesis) relative to I.
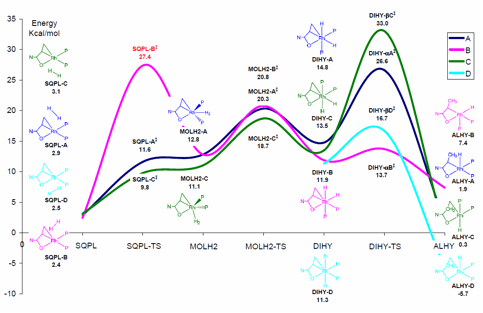
Figure 1: Energy profile related to the acrylamide hydrogenation assisted by Rh(I) catalyst. Energies reported in kcal/mol and referred to complex I + H2 including thermal contribution to the Gibbs free energy.
Geometries optimized for SQPL-A (2.9), B (2.4), C (3.1), and D (2.5) show hybrid orientations for all four pathways with each of these species located on a very flat region of the potential energy surface (PES). The SQPL-TS-A (11.6) and C (9.8) were identified and characterized through vibrational analysis. Several attempts have been made to locate equivalent transition states for the B and D pathway. So far, the unique transition state SQPL-TS being characterized for the B pathway shows an inaccessible energy barrier but is still under investigation. Any attempt to find SQPL-TS for the D pathway led to an isomerization from the D to the C orientation.
In the case of MOLH2 complex, only the A (12.8) and C (11.1) geometries were optimized, leading to a distorted trigonal bipyramid. The transition states MOLH2-TS lead to the dihydride complexes A (20.3), B (20.8), and C (18.7).
It was possible to characterize DIHY-A (14.8), B (11.9), C (13.5), and D (11.3). The migratory insertion occurs through the transition state DIHY-TS, which is the stereoselectivity determining step in the enamide reduction. Because of the hydrogen orientation, the four DIHYs go through DIHY-TS-aA (26.6), DIHY-TS-aB (13.7), DIHY-TS-bC (33.0), and DIHY-TS-bD (16.7).
In light of these results, it is not possible to exclude a priori the B and D pathways, which means that the DIHY-TS may not be the rate or stereoselectivity determining step. In other words, the reaction may occur by the C (or equivalently the A) pathway until the DIHY forms. Then, an isomerization process converts DIHY-C to DIHY-B (or DIHY-D). At this stage the reaction may continue to the ALHY-B (or ALHY-D) through a b transfer.
At this time, the insights that we have garnered about the mechanism of this reaction are being used to create a molecular mechanics force field for the stereoselectivity determining transition states. With this force field in hand, it will be possible to perform a fast computational virtual screen for either previously known or newly designed ligands affording high enantioselectivity by comparing the energy differences between the diastereomeric transition states. In this way, it will be possible to identify or design promising ligands for experimental consideration.



