Reports: AC9
46928-AC9 Atomic Scale Simulation of Many Body Forces in Colloidal Nanoparticle Suspensions
A quantitative understanding the forces between colloidal nanoparticles would benefit numerous applications in sensing, nanoelectronics, composite materials, nanofluids, optics, and catalysis that could exploit the unique properties associated with their small size. In applications involving crystallization, the aggregation of colloidal nanoparticles can be important and recent experiments have documented the phenomenon of oriented attachment, in which nanocrystals apparently approach one another and merge along specific crystallographic directions to form twinned or single-crystal structures [1-3]. The ability to direct crystallization through oriented attachment is an exciting prospect that could allow for the creation of new nanostructures with well-defined sizes and shapes. To realize the full potential of oriented attachment for achieving the controlled synthesis of a broad range of nanomaterials, insight into its origins, which are not currently clear in all cases, would be beneficial.
Recent progress in our group has been in two areas:
A. MD Simulation of the Aggregation of Titanium Dioxide Nanocrystals: Preferential Alignment
We used classical MD simulations to study the aggregation of various titanium dioxide (anatase) nanocrystals in vacuum. We consider charge-neutral nanocrystals with variations of the truncated tetragonal bipyramidal Wulff shape. Nanocrystalline anatase has been experimentally observed to possess approximate Wulff shapes [3]. The model nanocrystals are shown in Fig. 1, where we see that in addition to Wulff shapes, we consider asymmetric nanocrystals, which mimic possible off-Wulff shapes that could occur during crystal growth. The asymmetric nanoparticles possess permanent dipole moments, while the symmetric nanocrystals do not. Thus, we studied the effect of dipole-dipole interactions, which have been proposed to play a role in oriented attachment [1,2].
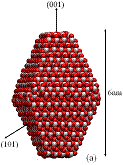
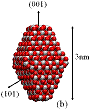
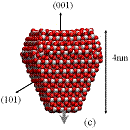
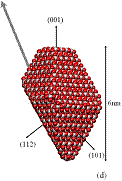

Figure 1: We studied (a) large crystals with Wulff shapes; (b) small crystals with Wulff shapes; (c), (d), (e) truncations of the Wulff shape, in which the relative magnitudes and directions of the dipole moments are indicated by gray arrows. Oxygen atoms are red (dark) and titanium atoms are white (light).
All the particles studied exhibit a strong tendency to aggregate with certain preferred orientations in a ``hinge'' mechanism. Our simulations indicate that electrostatic forces between under-coordinated atoms on the edges between nanocrystal facets drive this phenomenon. We find that aggregation rarely occurs along the direction of the dipoles - even when the permanent dipole moment is as large as 250 Debye. Although the dipole-dipole interaction is the leading term in the multipole expansion of the electrostatic potential for neutral particles, higher order multipole moments (e.g., quadrupole, octupole, etc.) can also contribute to the electrostatic potential. Here, these high-order multipoles dominate the electrostatic potential at short separations and they are created by regions of positive and negative charge associated with under-coordinated surface atoms, as shown in Fig. 2. The observed mechanism for preferential alignment may be a driving force for oriented attachment and the growth of anisotropic structures during crystallization.
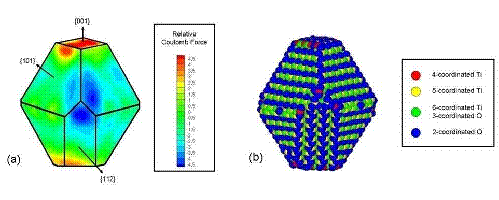
Figure 2: (a) Projected map of the electrostatic potential in planes parallel to the nanocrystal facets on the particle shown in Fig. 1(e). The coordination numbers of the surface atoms are indicated in (b). Nanoparticle aggregation occurs between the red (positively charged) and blue (negatively charged) regions shown in (a).
A. MD Simulation of the Directed Alignment of Au Nanocrystals in Alkane Solvent: Solvation and van der Waals Forces
We used MD simulations to study the approach of icosahedral gold nanoparticles in hexane solvent, where we find an interesting interplay between solvent-mediated forces and van der Waals attraction. In the absence of solvent, the nanoparticles approach one another in a facet-to-facet orientation, due to van der Waals forces, as shown in Fig. 3(a). Hexane solvent tends to decorate the nanoparticle facets, leaving the vertices and edges bare, so that in solvent, the nanoparticles approach each other in a vertex-to-vertex fashion [Fig. 3(b)]. This study shows the critical role of solvent in achieving nanoparticle alignment.
(a)
(b)
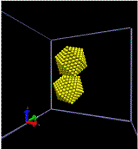

Figure 3: Snapshots from MD simulations of nanoparticle aggregation (a) in vacuum and (b) in solvent. For clarity, hexane solvent molecules are not shown to scale.
Future Plans
Work is continuing on the projects described above. We are currently simulating TiO2 nanoparticles in an aqueous environment to understand the role of hydroxyl groups and hydration on nanoparticle alignment and aggregation. These simulations will be directly relevant to the experimental studies of the crystallization of colloidal anatase, where oriented attachment was first discovered [3]. Additionally, we are probing the long-time restructuring of the anatase nanoparticles after they aggregate using accelerated MD simulations. In our MD studies to date, the aggregates are polycrystalline and, hence, are not the monocrystals observed in experiment [3]. By using accelerated MD simulations, we will see if monocrystals form over times that are short on the experimental scale, but long over the (ns) time scale of regular MD. These studies will provide the first insight into long-time aspects of the sintering process.
For the gold/hexane system, we are looking at two different angles. First, we are simulating the aggregation of the nanoparticles in solvent. Our simulations show that these particles should have vertex-to-vertex alignment when their closest atoms are just 1 Å apart and we would like to know if the particles actually aggregate vertex-to-vertex. Strong bonding and van der Waals interactions both favor aggregation in the facet-to-facet configuration and vertex-to-vertex aggregation would be favored by kinetics and solvation forces. We are also simulating the interactions between a long (anisotropic) nanoparticle and a compact icosahedron. The hypothesis is that solvation forces are the most repulsive for large, smooth surfaces, which allow solvent molecules to achieve good ordering. Anisotropic nanoparticles will have long, smooth sides, where solvent can order well, and small ends, where solvent cannot order. Thus, we expect a small, uniform nanoparticle to be attracted to the end more than to the sides. These simulations will provide insight into the formation of anisotropic structures during crystallization.
References
1. Niederberger, M.; Colfen, H. Phys. Chem. Chem. Phys. 2006, 8, 3271.
2. Zhang, Q.; Liu, S.-J.; Yu, S.-H. J. Mater. Chem. 2009, 19, 191.
3. Penn, R. L.; Banfield, J. F. Geochim. Cosmochim. Acta 1999, 63, 1549.



