Reports: AC6
45988-AC6 Understanding Chemical Bond Dynamics in Solution using Femtosecond Spectroscopy and Nonadiabatic Mixed Quantum/Classical Simulations
On the theoretical side of the project, graduate student Will Glover has completed his development of the new method for performing CISD calculations for electronic structure during mixed quantum/classical dynamics, and has submitted two papers related to this work, the first detailing the method and the second detailing the results for the bond dissociation dynamics of the sodium dimer in liquid Ar. Some of the ideas underlying the method and the principle results of this work were summarized in last year's report; the TOC graphic shows the significant difference in solvation structure that results when a quantum description is used for the dimer bond instead of a classical description. Since Will will be graduating in September 2009, the theoretical part of the project will be picked up by a new graduate student, Argyris Kahros. Argyris has been focusing on the development of new pseudopotentials that depend on the length of the sodium dimer bond. This development is critical for the accurate simulation of bond-breaking/formation dynamics because traditional pseudopotentials suffer from what is known as the 'frozen-core' approximation: the pseudopotential is calculated for a fixed electronic structure, and thus cannot respond to changes in electronic structure that occur during the course of the simulation. For the sodium dimer, the pseudopotential needed to calculate the electronic structure of the isolated atoms near the dissociation limit is quite a bit different than that needed to calculate the nature of the diatomic bond near equilibrium. Our goal is to go beyond the frozen core approximation and produce non-frozen-core pseudopotentials that can be used in quantum simulations of chemical bond breaking. Argyris is planning on calculating how the pseudopotential for a sodium dimer needs to change along the dissociation coordinate. He will then implement this coordinate-dependent pseudopotential into a molecular simulation, and ultimately to compare the results he gets to a full quantum calculation. If the coordinate-dependent pseudopotential produces accurate dynamics, it would open an entire realm of new possible quantum simulations, where the quantum dynamics of large systems can be calculated because the potentials change on the fly as necessary to correctly account for the dynamics of the implicitly-treated electrons.
On the experimental side of the project, graduate student Stephanie Doan has been hampered in making further progress because of difficulties with sample handling: the alkali hydride molecules we are studying are highly air-sensitive, but the walls of the air-tight sample cells we use in our experiments produce pump-probe artifacts that obscure the spectroscopy of interest. To get around this problem, Stephanie has been focusing on the development of air-free solvent jets: the idea is to stream the sample in a liquid jet, but keep the jet in an air-free environment to avoid problems with the high reactivity of the hydrides. Stephanie now has such jets working, and as a first test, she has explored the photodecarbonylation of DPCP to DPA (see Fig. 1). In solution, photoexcitation of an isolated DPCP molecule at ~300 nm leads to formation of DPA with unit quantum yield.[1] In the solid state, however, this same reaction has a quantum yield greater than 3: there is a 'quantum chain reaction' whereby excess energy released from the first photoconversion triggers the reaction of neighboring molecules.[2] In collaboration with our UCLA colleague Miguel Garcia-Garibay and his group, we have studied this photoconversion process in a nanocrystalline suspension of DPCP flowed through our air-free liquid jet set-up.[3] The transient absorption dynamics are shown in the left panel of Figure 1. Since DPA is formed in its electronic excited state, we show the same experiment on a sample of DPA in the right-panel of Figure 1. The transient absorption feature seen near 500 nm in the DPCP signal is the clear signature of the excited intermediate that causes the quantum chain reaction, and its decay on a ~1.5 ps time scale provides the first direct measurement of such processes. We will continue to explore both this reaction and the photodissociation of KH in liquid THF in future experiments.
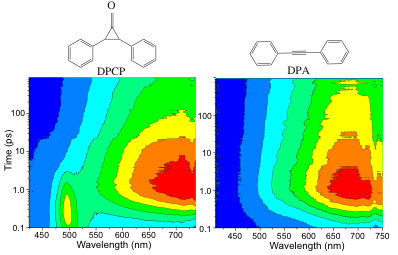
Figure 1: Ultrafast transient absorption dynamics of nanocrystalline suspensions of DPCP (left) and DPA (right) flowed through a liquid cyclohexane jet following 300-nm excitation. The color scale runs from red (max transient absorption) to blue (no transient absorption).
[1] S. Takeuchi and T. Tahara, J. Chem. Phys. 120, 4768 (2004).
[2] G. Kuzmanich, A. Natarajan, K. K. Chin, M. Veerman, C. J. Mortko and M. A. Garcia-Garibay, J. Am. Chem. Soc. 130, 1140 (2008).
[3] G. Kuzmanich, M. N. Gard and M. A. Garcia-Garibay, J. Am. Chem. Soc. 131, 11606 (2009).



