Reports: AC3
45941-AC3 Harnessing Metal-Ligand Cooperative Redox Reactivity for Bond Functionalization Strategies
Introduction
The fundamental goal of this project was to develop a new platform for multi-electron reactivity comprising redox-active ligands and early transition metals. In this work we have focused on a tridentate amido-bis(phenolate) ligand (ONO); the three oxidation states of the ONO ligand are shown below.
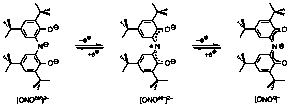 Preparation of zirconium complexes of the [ONO] ligand
Preparation of zirconium complexes of the [ONO] ligand
Both mono- and dihalide complexes of zirconium were prepared through dehydrohalogenation reactions between [ONOcat]H3 and ZrCl4. Various bases were used to prepare the dichloride, [ON(H)Ocat]ZrCl2(solv) (solv = Et2O, THF, pyridine) (1). Complex 1 is a stable species that can be isolated and stored for long periods of time. Alternatively, we have prepared [ONOcat]ZrCl(THF)2 (2) by triple deprotonation of [ONOcat]H3 and reaction with ZrCl4 in THF. Figure 1 shows the structure of 2 as determined by single-crystal X-ray diffraction. Sodium azide reacted with 2 to form a new zirconium azide complex [ONOcat]Zr(N3)L2 (3, L = THF, py), which was isolated in modest yield. Figure 2 shows the new zirconium azide, crystallized with one coordinated pyridine and one coordinated THF molecule.
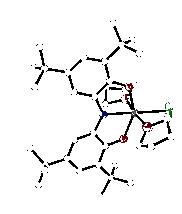
Figure 1. Molecular structure of [ONOcat]ZrCl(THF)2 (2) as determined by X-ray diffraction.
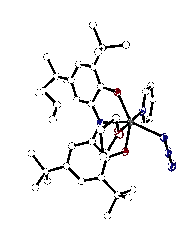
Figure 2. Molecular structure of [ONOcat]Zr(N3)(THF)(py) (3) as determined by X-ray diffraction. Oxidation studies of the [ONO]Zr platform
The goal of this research program was to develop the multi-electron redox reactivity of the ONO ligand platform coordinated to electrophilic transition metals. To benchmark the redox reactivity of the ONO ligand coordinated to zirconium, halogen oxidation reactions were carried out. The addition of 0.5 equiv of PhICl2 to solutions of 2 resulted in an immediate color change from yellow to dark blue. Paramagnetically-broadened NMR spectra of the product indicated one-electron oxidation of the ONO ligand to the semiquinonate oxidation state and accordingly the EPR spectrum of the complex showed an S = ½ spin system with the unpaired electron localized on the ligand. Based on the available spectroscopic data, and similar reactions carried out on analogous tantalum complexes, the one electron oxidized product is formulated as [ONOsq¥]ZrCl2(py) (4). Addition of one equivalent of PhICl2 to 2, resulted in two-electron oxidation to afford the trichloride species [ONOq]ZrCl3 (5). The complex was characterized by NMR spectroscopy with the most diagnostic features being a pair of proton resonances at 7.33 and 7.29 ppm for the aromatic backbone of the oxidized ONO ligand.
An intriguing aspect of metal azide complexes is their propensity to expel N2 to generate a metal nitride under heating or upon irradiation with visible light. In order for such a reaction to occur, the metal center must provide two electrons to aid in formation of the metal-nitrogen triple bond. In the case of 3, which is formally a d0 zirconium(IV) complex, the metal center cannot provide the requisite electrons for such a reaction; however, the redox properties of the ONO ligand, established above, suggested that the reaction may be accessible through the use of ligand-based electrons. Along these lines, complex 3, which affords deep red solutions in MeCN, changes to violet upon heating for several days. The 1H NMR spectrum of the product showed resonances at 7.26 and 7.24 ppm, diagnostic for the [ONOq]– form of the ligand. Toeppler pump experiments indicate the loss of one equivalent of N2 during the reaction and solution molecular weight measurements are consistent with the formation of a terminal zirconium nitride, [ONOq]Zr(≡N)(py)2 (6). To our knowledge this result is the first example of the formation of a zirconium(IV) nitride from a zirconium(IV) azide.
Two electron reactivity was realized from complex 1 upon treatment with stoichiometric oxygen. Complex 1 contained a reduced ONO ligand with a single proton on the bridging nitrogen. In its protonated form, the ONO ligand platform displays significantly reduced redox reactivity; however, 1 reacts immediately with molecular oxygen to form a new zirconium-hydroxo dimer, {[ONOq]ZrCl2(μ-OH)}2 (7) in nearly quantitative yields. As is typical, the 1H NMR spectrum of 7 displays two resonances for the aromatic backbone of the oxidized ligand at 7.38 and 7.35 ppm. Moreover, a diagnostic stretch at 2960 cm–1 in the IR spectrum indicated the presence of bridging hydroxo ligands. Figure 3 shows the molecular structure of 7 as determined by single-crystal X-ray diffraction. Each metal center is roughly pentagonal bipyramidal with axial chloride ligands and the hydroxide ligands in the same plane as the oxidized ONO ligand. Consistent with the formal d0 electron count at each metal center, the Zr–Zr distance is long at 3.55 .
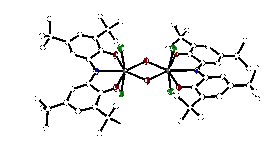
Figure 3. Molecular structure of {[ONOq]ZrCl2(μ-OH)}2 (7) as determined by X-ray diffraction. Conclusions
The work completed during this funding period established the viability of the ONO ligand as a redox-active platform for electrophilic early transition metals such as zirconium. Figure 4 summarizes the new reactivity discovered for the zirconium-ONO platform. Multi-electron reactions including oxidative addition, nitrogen atom transfer, and oxygen activation have all been observed. In the case of O2 activation, two ONO ligands work together to provide the four electrons required to completely cleave O2 and also serve as the source of the two protons to generate the hydroxo ligands. As such, this work shows that redox-active ligands such as this one can be incorporated to work as reservoirs for both protons and electrons in small-molecule activation reactions. Given the importance of coupled proton-electron transfer reactions in small-molecule activation, this result highlights the potential impact of redox-active ligands in the design of new metal catalysts for small molecule activation.
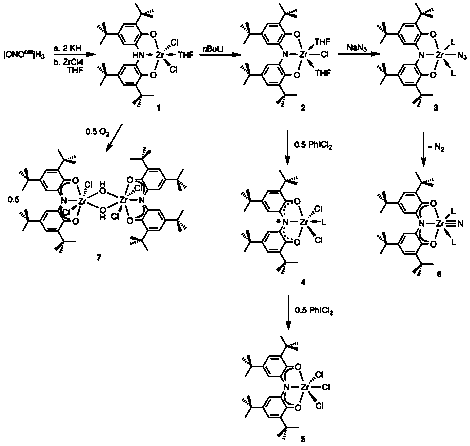
Figure 4. Summary of reactivity for the zirconium-ONO platform.



