Reports: B6
47856-B6 The Development of Accelerated Molecular Dynamics for Complex Gas-Phase Reactive Systems
Significant progress has been made in the writing of a computational tool for performing molecular dynamic simulations of complex chemical reactions in the gas-phase. A suite of programs called Accelerated Molecular Dynamics with Chemistry (AMolDC) has been written, tested, and employed in order to perform multilevel QM/MM simulations. The properties of this new method were investigated. The method is formulated to give a time-dependent, multilevel representation of the total potential that is derived from spatially-resolved quantum mechanical (QM) regions. A 110 atom system was used to demonstrate the continuity and energy conserving properties of the method. The effect of a discontinuous total potential upon the kinetic energy of the system was examined. The discontinuities in the magnitude of atomic force vectors due to changing the electronic structure during the simulation were examined, as well as the effect that these discontinuities have upon the atomic kinetic energies. The method, while not conserving total energy, does yield canonical (NVT) simulations. The time-reversibility property of the simulation with an extremely discontinuous total potential was also examined in addition to the computational scaling and savings associated with the formation of the spatially-resolved, time-dependent groups.
 The 110 atom gas-phase system employed for
examining the continuity and computational properties of the time-dependent,
multilevel QM/MM simulations consisted of individual C2H4,
CO2, and CO molecules.
When these C2H4, CO2, and CO molecules
were isolated, they were treated at the MP2/McLean and Chandler (MC), SVWN/6-31G,
and RHF/STO-3G levels of theory, respectively. For groups mixed with combinations of molecular systems, the
following rules were employed: combinations of molecules containing all three
CO, CO2, and C2H4 species, the RHF/STO-3G
level of theory was employed; for CO and C2H4 only, the
SVWN/6-31G level of theory was employed; for any combination of CO2
and C2H4 with fewer than 20 atoms, the MP2/MC level of
theory was used; for any combination of CO2 and C2H4
with more than 20 atoms, the RHF/STO-3G level of theory was employed.
The 110 atom gas-phase system employed for
examining the continuity and computational properties of the time-dependent,
multilevel QM/MM simulations consisted of individual C2H4,
CO2, and CO molecules.
When these C2H4, CO2, and CO molecules
were isolated, they were treated at the MP2/McLean and Chandler (MC), SVWN/6-31G,
and RHF/STO-3G levels of theory, respectively. For groups mixed with combinations of molecular systems, the
following rules were employed: combinations of molecules containing all three
CO, CO2, and C2H4 species, the RHF/STO-3G
level of theory was employed; for CO and C2H4 only, the
SVWN/6-31G level of theory was employed; for any combination of CO2
and C2H4 with fewer than 20 atoms, the MP2/MC level of
theory was used; for any combination of CO2 and C2H4
with more than 20 atoms, the RHF/STO-3G level of theory was employed.
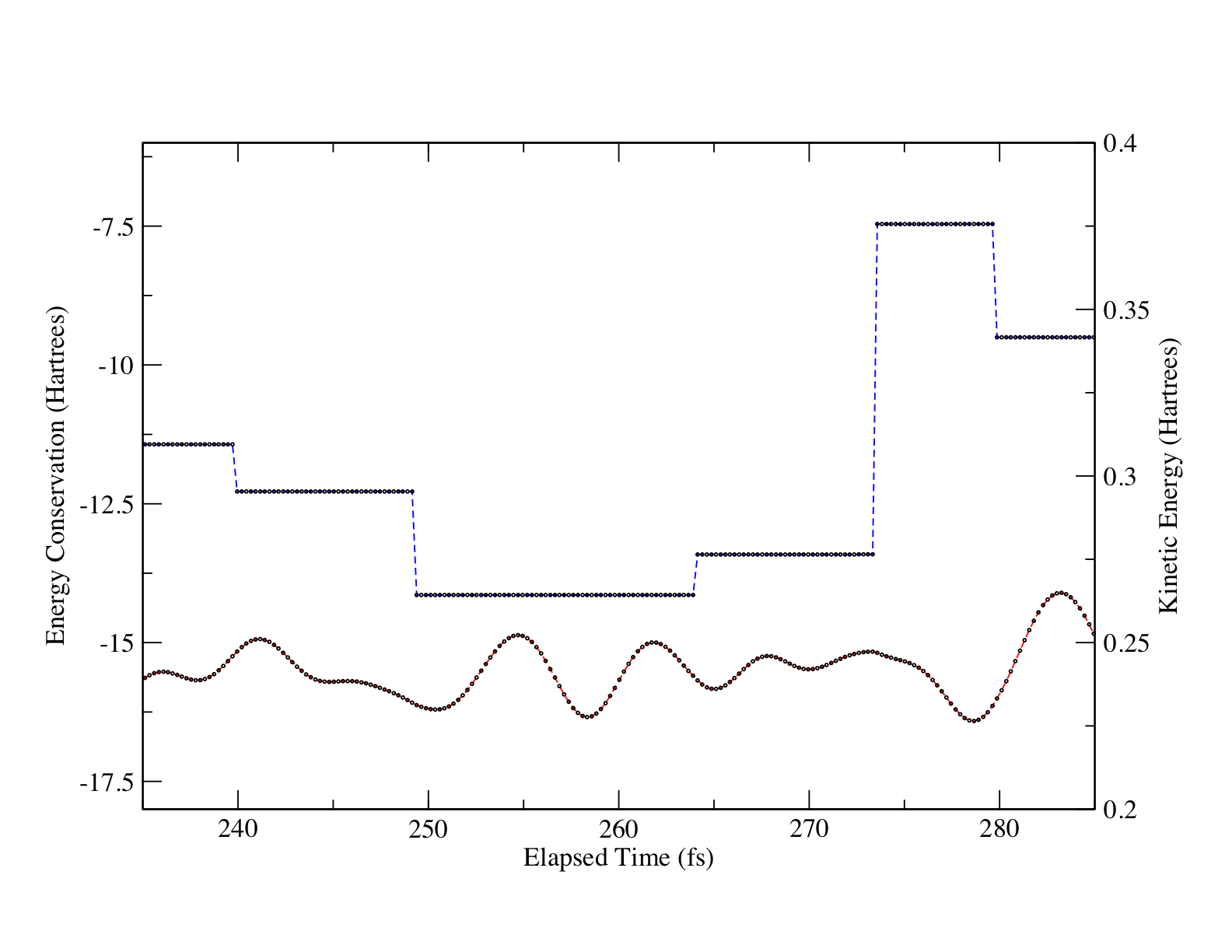
Figure 1 shows the total energy and kinetic energy of the 110 atom simulation during a small time window that had 5 significant discontinuities in the total potential due to the changing of groups and levels of electronic structure theory to describe these groups. From Figure 1 it can be seen that, although the total potential is radically discontinuous (by over 5 Hartrees at one point), the kinetic energy is smooth and continuous throughout. A continuous kinetic energy leads to canonical (NVT) simulations.
In order to examine the effect of discontinuities in the intragroup potential due to changes in the level of theory (i.e., going between MP2/MC, SVWN/6-31G, and RHF/STO-3G levels of theory), the magnitude of the atomic force vectors were calculated for atoms undergoing a change of theory. Discontinuities of as large as 0.0506 Hartree/bohr were seen in these atomic force vectors due to changes of theory. Despite these significant discontinuties in the atomic forces, the atomic kinetic energies moved smoothly across the areas of discontinuity.
Time-reversibility of the simulations were examined over the region seen in Figure 1, where the total potential is radically discontinuous. The root mean square (rms) error in the atomic coordinates rises slowly and in a very linear manner to reach a maximum value of 3.635×10-4 after 60 fs and there is not additional error where there are changes in groups and discontinuities in the total potential.
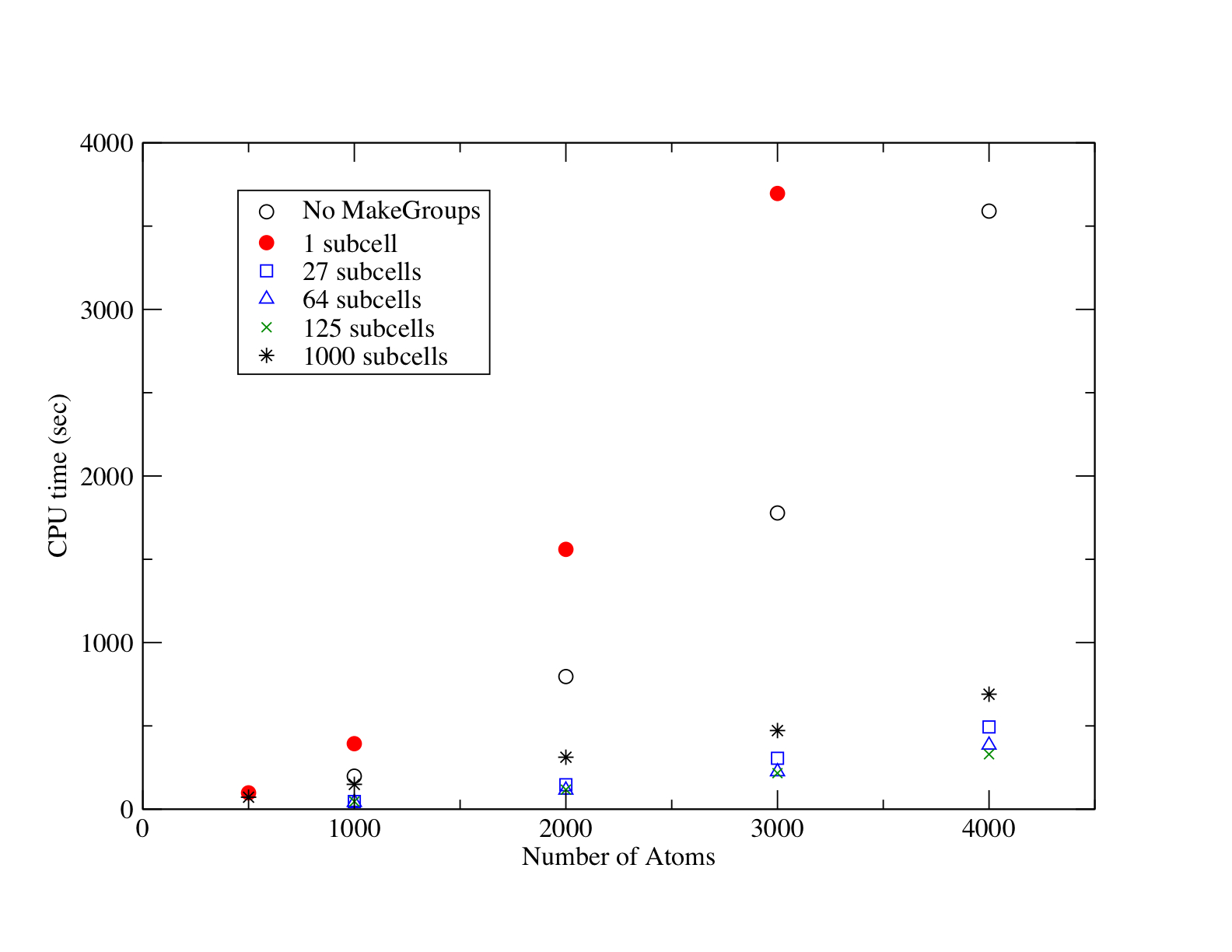
The MakeGroups module within AMolDC has been programmed in order to accomplish the task of making the spatially-resolved groups at each time step in a simulation. In order to investigate the computational scaling of the MakeGroups module, simulations of N2 at 300K were performed in which the nitrogens were randomly placed within a simulation cell and propagated for 500 time steps using a simple Morse potential for all internuclear distances. As the number of N2 molecules was increased, the simulation cell size was adjusted to yield an ideal gas pressure of ~90 atm at 300K. Shown in Figure 2 are the total simulation times without using the MakeGroups module (open circles) and using the MakeGroups module with 1 subcell (red filled circles), 27 subcells (open boxes), 64 subcells (open triangles), 125 subcells (×), and 1000 subcells (astericks). One can see from this figure the O(N2) scaling when not using MakeGroups (calculating all interatomic distances) and when using MakeGroups with only 1 subcell (the whole simulation cell). The computational overhead for the MakeGroups module is 1.45 times that of calculating all internuclear distances as measured by the coefficient of the leading O(N2) term over the same simulation. However, when the simulation cell is divided into a distribution of 3×3×3 link-listed subcells, the computational time is greatly reduced and the computational expense goes from O(N2) scaling to O(N) scaling. There is little computational difference (relative to the same simulation without MakeGroups) between a 3×3×3, a 4×4×4, and a 5×5×5 division of link-listed subcells. Saturation of the simulation cell with subcells, as given most closesly by the 10×10×10 division of link-listed subcells (astericks), shows almost exactly linear scaling in the number of atoms. Even though the computational cost of MakeGroups is insignificant relative to high-level QM calculations, what is demonstrated in Figure 2 is that, because the total potential is assembled from time-dependent groups, one has the ability to reduce the total number of QM calculations in order to yield overall linear scaling with system size.
The results of all of these investigations have been submitted to Journal of Chemical Theory and Computation on August 24th, 2009.



