Reports: AC1
47616-AC1 Formation of Functional Amino Acids via Three-Component Coupling: An Unusual Addition Mode to the Nitrogen of alpha-Iminoesters
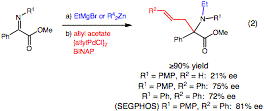
alpha-Amino acids and their derivatives are vital synthetic building blocks in organic synthesis and play an integral role in pharmaceutical research. Therefore, the synthesis of both natural and non-natural alpha -amino acids continues to be a subject of intense study. In particular, the formation of alpha,alpha-disubstituted alpha -amino acids is difficult due to the quaternary center even though such compounds have useful properties. For example, when incorporated into peptides, alpha,alpha -disubstituted alpha-amino acids confer increased stability under physiological conditions and stabilize secondary structure motifs. They also have relevance in natural product total synthesis. Herein, we report that alpha-iminoesters engage in an umpolung addition with organometallic agents providing a three component coupling route toward alpha,alpha-disubstituted alpha-amino acids.
Our laboratory has discovered processes for the asymmetric addition of diorganozinc reagents to aldehydes, alpha-ketoesters, and alpha-aldiminoesters. During exploration of the related alpha-ketiminoesters, we observed an unusual reaction pattern. In addition to the expected C-alkylation and reduction products, an N-alkylation product was observed (Eq. 1). Further exploration of this process under the aegis of this PRF grant revealed that is possible to form any of the three products in Eq. 1 selectively. Electron-withdrawing groups on nitrogen tend to favor reduction. With N-aryl substitution, the more reactive organolithiums favor C-alkylation while the Grignard, aluminum, and zinc reagents bias the reaction toward N-alkylation.
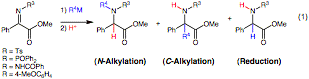
The tetra-substituted enolate resulting from the N-alkylation is of considerable interest. Both ends of the double bond in the resultant enolate are potentially nucleophilic and we found that the reaction of the ketene acetal anion predominated (Figure 1). A myriad of complex structures can arise such a transformation when different electrophiles are employed leading to the synthesis of alpha,alpha-disubstituted alpha-amino acids (Figure 1).
Optimization of the N-alkylation reaction revealed that alkyl magnesium bromides in THF were superior. Upon protonation of the enolate adduct, the products from EtMgBr and n-pentylMgBr could be obtained in 84 and 85% yield, respectively. Furthermore, the enolate could readily be exploited to achieve a one-pot three component coupling reaction using a range of electrophiles including aldehydes, imines, nitroalkenes, acyl cyanides, and alkyl halides (Figure 1).
Figure 1. Sample products from the three-component coupling with various electrophiles.
When benzaldehyde was employed as the electrophile high conversion (~80%) to the three component product as a single diastereomer was seen. This result is surprising given that Shimizu and coworkers report self-addition (addition of the in situ generated enolate to the a-iminoester substrate) in a related system. To our knowledge, this represents the first example of three unique components being combined in this type of alpha-iminoester N-alkylation/electrophilic C-alkylation. Shimizu and coworkers have reported a related, but considerably more complex, process in which the initial enolate adduct is oxidized with benzoyl peroxide and the resultant cation reacts with allyltributyltin.
Additional aldehydes reacted with high diastereoselection with representative Grignard reagents (R4 = Et, n-pentyl) providing the three component coupling products in very good yield (Figure 1). The N-tosyl imine also proved to be an effective electrophile furnishing a single diastereomeric product (Figure 1). The syn diamine relative stereochemistry was verified from the crystallographic structure and is in accord with a chair-like transition state commencing from the Z enolate.
Initial attempts to use alkyl halide electrophiles provided very low yields of the three component coupling products (<10%). We discovered that DMF as a solvent greatly increased the yield in this instance. For example, reaction with BnBr or EtI afforded 72% and 66% yield. Furthermore, tert-butyl bromoacetate provided in 48% yield (Figure 1). Conjugate acceptors were also effective electrophiles. For example, trans-b-nitrostyrene furnished the three component products in good yields. While acid chlorides gave poor yields, acyl cyanides such as pyruvonitrile were particularly effective, furnishing the product in good yields (Figure 1). Notably, different a-iminoesters with various ester groups, different aryl substitution, and even different N-substitution could be employed.
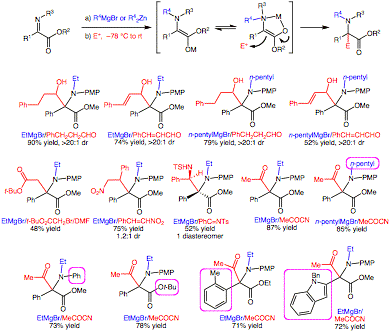
The opportunity to construct heterocycles utilizing this method is considerable. For example, a Grignard reagent containing a terminal chloride was employed (Figure 2). After Grignard addition to the nitrogen, C-alkylation with the halide did not proceed until DMF was added to the reaction mixture. This protocol permits formation of unusual amino acids derived from piperidines and azepanes. Also, removal of the PMP group was straightforward with ceric ammonium nitrate (CAN).
Figure 2. Tethered nucleophiles/electrophiles.
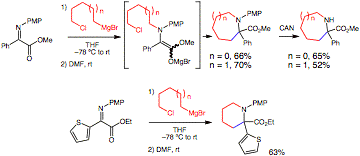
We have also discovered that it is possible to trap the enolate resulting from N-alkylation of the a-iminoester with an allyl acetate (Eq. 2). However, in this case, no reaction occurrs unless a palladium catalyst is employed. Since these conditions of ligand-accelerated catalysis are ideal for asymmetric catalysis, the transformation has been examined with chiral palladium complexes. To date, the complex of allylpalladium chloride with SEGPHOS has been the most effective providing the chiral alpha, alpha-disubstituted amino acid product with 81% ee. Future efforts will focus on further ligand screening and modifying the substrate groups (PMP and ester) to improve the selectivity.
In conclusion, the rapid construction of alpha, alpha-disubstituted alpha-amino acid derivatives can be achieved in high yields from alpha-iminoesters. The reaction proceeds through an umpolung addition of an organometallic reagent to the nitrogen. The resultant enolate reacts with a variety of electrophiles to form a quaternary center. Furthermore, high diastereoselection occurred in the three component coupling reactions with aldehydes and imines. This methodology provides straightforward access to a variety of pharmacologically and synthetically useful products.



